o algoritmo básico de busca de alinhamento Local (BLAST) está no coração de um conjunto livre de recursos online disponíveis através do National Center for Biotechnology Information (NCBI). Enquanto a maioria dos pesquisadores estão cientes do BLAST como uma ferramenta de alinhamento de seqüências, o pacote BLAST do NCBI oferece muito mais! Eu vou cobrir em profundidade como usar esses recursos para localizar polimorfismos de nucleótidos únicos (SNPs) em um gene; projetar primers com primer-BLAST; e validar alvos de primer.
Ponta 1: Como encontrar SNPs
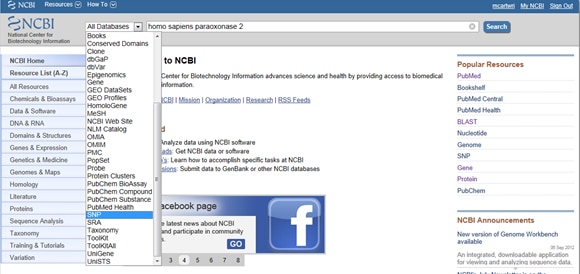
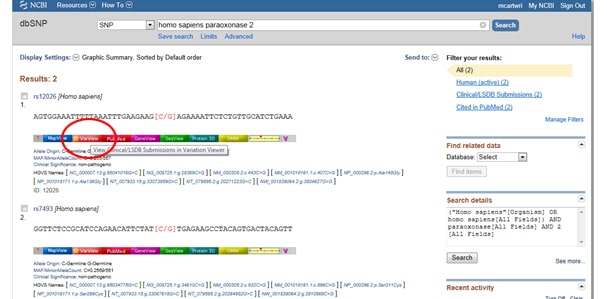
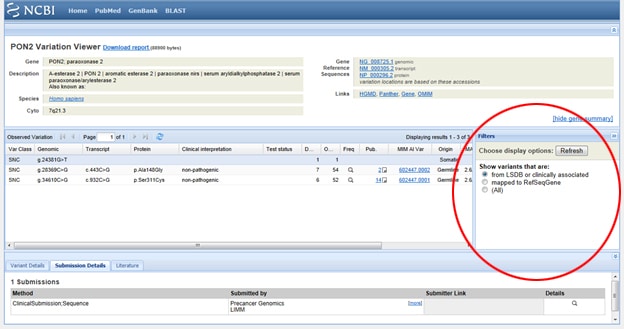
dada a importância dos SNPs tanto na doença quanto na pesquisa, NCBI fornece ferramentas para a colagem dos SNPs relatados por um gene. Para encontrar SNPs, Comece na página inicial do NCBI e digite seu gene de interesse na barra de pesquisa. Selecione SNP a partir de Todos os Bancos de dados no menu suspenso à esquerda da barra de pesquisa, como mostrado abaixo:
dica dois: Como desenhar os iniciadores
NCBI fornece Primer-BLAST para desenhar automaticamente os iniciadores com base numa sequência de consulta. Para começar a desenhar os iniciadores, vá para a homepage da explosão e vá para a opção Primer-BLAST sob explosão especializada. Digite sua sequência de destino por corte – e-colar ou, se estiver listado nas bases de dados do NCBI, como um número de adesão. Eu cobrir algumas opções de personalização abaixo, mas neste ponto, você pode gerar primers sem fazer qualquer personalização adicional!
intervalo: à direita do campo para introduzir a sua sequência, poderá indicar o intervalo EXACTO (numerado de 5′ a 3′, desde o início da sua sequência) do alvo que será considerado para desenhar os iniciadores para a frente e para trás.
Use a minha própria primer para a frente (5′->3 ‘ Na Linha plus): seleccione isto se já tiver desenhado os seus iniciadores e quiser que a Primer-BLAST forneça alguma análise (por exemplo, Tm) sobre eles.
PCR Tamanho do produto: Defina aqui a gama de comprimentos aceitáveis dos produtos PCR.
# de iniciadores a retornar: isto define o seu número preferido de conjuntos candidatos de iniciadores a considerar. Note que não é uma garantia, especialmente se seus parâmetros são muito rigorosos ou sem sentido (por exemplo, você especificou um produto sob o tamanho do produto PCR que não pode ser mais de 500 bp, mas sob o intervalo você só quer considerar os iniciadores com mais de 1 KiB de diferença).temperaturas de fusão dos iniciadores: Isto permite-lhe especificar o seu Tm (para uma actualização rápida sobre a temperatura de fusão, verifique as nossas dicas para o qPCR e o design regular do primer PCR).
regulação da junção Exon: se quiser excluir o ADN genómico (em que os exons são divididos por intrões não codificadores), então configure isto como Primer, tem de abranger uma junção exon-exon.verificação da especificidade de
: A menos que queira que o Primer-BLAST devolva os primers que irão sair do alvo (geralmente não recomendado!), deixe este verificado e especificar o organismo que suas amostras estão vindo, bem como Qual banco de dados a usar, dependendo se você está visando mRNA, gDNA, etc. Ao permitir a verificação da especificidade, o Primer-BLAST excluirá os primers que podem amplificar algo fora da sequência de alvo.manuseamento da variante da unidade: se seleccionar esta opção-apenas viável se estiver a trabalhar com sequências de mRNA – então a Primer-BLAST não irá excluir pares de primer que possam amplificar múltiplas variantes de mRNA do seu alvo. Isto não significa, no entanto, que lhe dará pares de primeros que englobam todas as variantes de splice conhecidas! Estás apenas a afrouxar os teus critérios de alvo.
Uma vez introduzida a sua sequência e personalizada conforme necessário, desloque-se para o fundo da página e, depois de verificar usar uma nova vista gráfica, carregue em obter iniciadores. Isto irá retornar um mapa de onde os pares de iniciadores sugeridos irão amplificar o seu alvo, bem como análises sobre os iniciadores: seu comprimento, localização precisa, respectivo Tm, GC%, e pontuações refletindo auto-complementaridade (com 0.00 refletindo nenhuma complementação prevista).dica Três: como prever os alvos de iniciadores
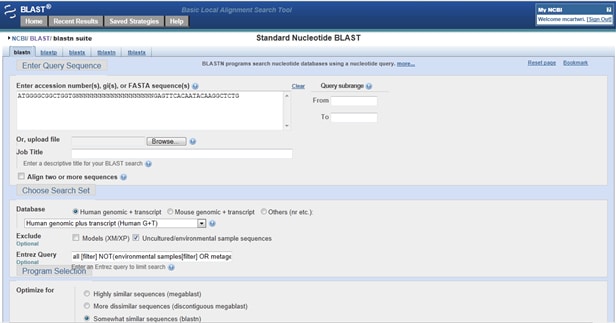
Como você pode verificar se os seus iniciadores acertaram alguma coisa fora do alvo? Vai para o Primer-BLAST. No campo de pesquisa, digite o seu primer avançado (5′ a 3′). Agora digite 20 NS seguidos para separar os iniciadores em alinhamentos individuais e não sobrepostos. Depois dos’ n’ s, introduza a sua primer inversa (também 5 ‘a 3’), Como mostrado abaixo:
References and Additional Resources:
Blast Tips. 2007. NCBI. <http://www.ncbi.nlm.nih.gov/feed/rss.cgi?ChanKey=blasttips>
Frequently Asked Questions. NCBI BLAST Help. <http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=FAQ>
Madden T. The BLAST Sequence Analysis Tool. 2003. <http://www.ncbi.nlm.nih.gov/books/NBK21097/>
Mount DW. Using the Basic Local Alignment Search Tool. 2004. Cold Spring Harbor Protocols. <http://cshprotocols.cshlp.org/content/2007/7/pdb.top17.full>
Wheeler D and Bhagwat M. BLAST QuickStart. 2007. Humana Press Inc. <http://www.ncbi.nlm.nih.gov/books/NBK1734/>
isso Tem ajudado você? Então, por favor, partilhe com a sua rede.