artigo de revisão
Leucemia para o clínico geral
Leukemia for the general practitioner
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
A chefe do serviço de Hematologia. Hospital Anjos do Pedregal. México, DF. E-mail: [email protected]
B Medicina Interna. Hospital Anjos do Pedregal. México. DF.
C Medicina Interna. Hospital Anjos do Pedregal. México. DF.
Recebido: 17 de outubro de 2011
Aceito: 07 Janeiro 2012
introdução
apesar dos grandes avanços moleculares e terapêuticos no estudo das leucemias, os aspectos básicos deste sofrimento ainda não são conhecidos de maneira clara pelo médico não hematologista, pelo que o objetivo deste trabalho é fornecer informação fundamental aos estudantes de medicina e médicos em geral, e que permita sobretudo obter o conhecimento geral das leucemias, seu diagnóstico oportuno e buscar a referência precoce com o hematologista.

DEFINIÇÃO
Leucemia é o termo usado para definir um grupo de doenças malignas do sangue. O diagnóstico precoce é essencial, pois permitirá que o paciente chegue cedo ao médico especialista em Hematologia, que conduzirá o processo diagnóstico e oferecerá o tratamento específico. Caracteriza-se por uma proliferação clonal, autônoma e anormal de células que dão origem ao resto das células normais do sangue (comportamento tumoral em geral).
O acima implica que uma célula precoce sofre uma mudança genética que fará com que uma clona (colônia) anormal de si mesma ocorra sem controle. Esta produção anormal é desordenada porque as células anormais se multiplicam em imagem e semelhança de si mesmas, pelo que ocupam paulatinamente o espaço da medula óssea normal e provocam anemia progressiva, sangramento anormal e predisposição às infecções. Por outro lado, quando as células anormais invadem outros tecidos, ocorrerá falha do funcionamento do órgão que se ocupa, por exemplo, a infiltração ao sistema nervoso central que ocorre na leucemia aguda linfoblástica (LAL) poderia manifestar-se com cefaleia, crises convulsivas, alterações motoras focalizadas, aumento da pressão intracraniana, e de não fazer o diagnóstico precoce e proporcionar o tratamento adequado, apresentará perda da função e consequências irreversíveis.
manifestações CLÍNICAS
O quadro clínico é diverso e dependerá do tipo de leucemia: aguda ou crônica, porém para as 2 existem manifestações clínicas inespecíficas (que ocorrem em qualquer doença):
1. Fadiga.
2. Cansaço fácil.
3. Fraqueza generalizada.
4. Desejo de ficar em repouso ou na cama.
5. Requer a ajuda de alguém para atender às suas necessidades pessoais.
as leucemias crônicas são de curso indolente e até 50% dos casos são descobertos em uma revisão clínica de rotina ou de laboratório em voluntários que se consideram saudáveis e migram para doar sangue, porém, conforme progride a doença, apresentam-se as manifestações inespecíficas mas agora são específicas (Tabela 1).

nas formas agudas, as manifestações específicas derivam da deficiência de alguma das linhas celulares:
1. Eritrócitos: síndrome anêmica cuja intensidade dependerá do grau de hipoxemia, independentemente do grau de anemia. Dispneia de esforços médios até a ortopedia.
2. Plaquetas: petéquias, equimoses nos membros, e em casos mais graves generalizados, hemorragia seca e úmida com epistaxe, gengivorragia, hematúria, juba ou hematoquesia. Muito grave no sistema nervoso central (SNC).
3. Leucócitos: febre, diaforese, infecções localizadas até uma franca septicemia (bactérias ou fungos). Eles ocorrem com neutropenia menor que 250 neutrófilos/mm3 totais.
síndrome infiltrativa: refere-se à implantação anormal em qualquer tecido, embora o frequente seja:
1. Hepatomegalia ou esplenomegalia (figura 5).
2. Adenomegalia (local ou generalizada).
3. Pele leucémica.
4. Dor óssea por expansão da medula óssea.
5. Tecidos moles (sarcoma granulocítico).
6. Testicular.
7. SNC.
8. Gengivas e qualquer site (figura 1).

distúrbios metabólicos: eles resultam da hiperprodução anormal de células malignas e aumento da apoptose.
1. Acidose.
2. Aumento da desidrogenase láctica (DHL).
3. Hipercaliemia.
4. Hiperuricemia.
5. Aumento da β2 microglobulina.

a evidência clínica predomina como a pedra angular da suspeita diagnostica das leucemias e de qualquer padecimento, mas o que segue é complementar o diagnóstico com o apoio do laboratório clínico na citometria hemática completa ou especial, o que quer dizer, a observação minuciosa do esfregaço de sangue periférico por pessoal técnico que tenha a preparação na identificação de células anormais e sobretudo leucémicas.

alterações laboratoriais que obrigam a uma revisão especial incluem:
1. Anemia (qualquer grau).
2. Leucopenia ou leucocitose (predominância de uma linha celular).
3. Trombocitopenia.
4. Combinações: bicitopenia ou pancitopenia.
deve tomar-se especial cuidado quando o laboratório reporta a presença de leucócitos ou linfócitos atípicos (podem ser blastos leucêmicos). É aconselhável solicitar a revisão de um especialista (figura 2).

o Aspirado de medula óssea é indispensável para no diagnóstico (figura 3) e requer 20% de blastos para estabelecer o critério de leucemia aguda em qualquer uma de suas variedades.

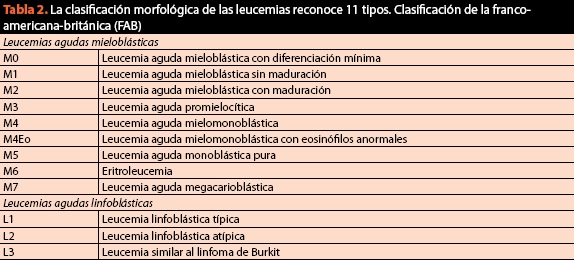
no mesmo procedimento devem-se obter amostras para a classificação final do padecimento e solicitar cariótipo e imunofenótipo, já que atualmente o critério citomorfológico é de vital importância mas já não é suficiente. Na (tabela 2) Se lista a classificação vigente e atual dos padecimentos malignos do sangue.
o tratamento destina-se a 2 aspectos importantes: o primeiro deles é o específico antileucêmico e baseia-se no uso de medicamentos de origem química que são conhecidos pelo nome de quimioterapia, cujo principal objetivo é erradicar, ou seja, eliminar todas as células leucêmicas do organismo. O segundo aspecto do tratamento é o apoio às complicações que os pacientes geralmente apresentam em sua admissão.
o tratamento é direcionado para 2 aspectos importantes: o primeiro deles é o específico antileucêmico e baseia-se no uso de medicamentos de origem química que são conhecidos pelo nome de quimioterapia, cujo principal objetivo é erradicar, ou seja, eliminar todas as células leucêmicas do organismo.
o segundo aspecto do tratamento é o suporte para as complicações que geralmente apresentam os pacientes em sua internação como são:
1. Anemia.
2. Hemorragia anormal.
3. Infecções pulmonares e generalizadas, entre outras (figura 4).
4. Qualquer outra complicação adjacente que o paciente possa ter (co-morbidade), como afecções pré-existentes, por exemplo, diabetes, hipertensão, cardiopatias e outras doenças frequentes entre os pacientes que sofrem de leucemia.


portanto, é muito importante notar que o tratamento contra a leucemia é multidisciplinar, o que implica a participação de outros especialistas como apoio ao hematologista.
o tratamento antileucêmico também será diferente para os diferentes tipos de leucemia e para as formas agudas. É dividido em 3 fases:
1. Indução da remissão. O objetivo é chegar à remissão completa (RC), isto é, a normalização dos valores do sangue do paciente, a ausência de quaisquer sintomas ou sinais de que a leucemia persiste com infiltração. Durante o processo o paciente deverá ter um “estado livre de leucemia” na medula óssea e o futuro deverá ser a recuperação a uma hematopoiese normal, e infelizmente em outros casos, se recuperam com a doença, o que fala de leucemia resistente cujo prognóstico é péssimo. Este primeiro processo pode levar de 6 a 8 semanas para alcançar o rc.
2. Consolidação. Envolve o uso dos mesmos medicamentos que foram utilizados na indução ou na combinação de outros quimioterápicos, também com o propósito de seguir a erradicação de células malignas residuais que possam desenvolver resistência aos de primeira utilização.
3. Manutenção. Prefere-se manter o paciente sob o efeito de quimioterapia ante a possibilidade de atividade leucêmica incipiente e que com o tratamento mantenha efeito até desaparecer a doença.

até agora, para formas agudas, os critérios mais rigorosos para considerar RC tornam-se mais complexos, já que o mais aconselhável é procurar a remissão molecular, que envolve a busca da alteração cromossômica inicial, como ocorre no caso da leucemia promielocítica(M3-FAB) com t (15;17) inicial, deve-se procurar por estudos moleculares e citogenéticos específicos apesar de estar em RC, já que se persistir a translocação, então deve-se continuar o tratamento agressivo até a eliminação total da clona. Esta variedade de leucemia deve ser considerada como potencialmente curável e a qual é, da mais frequente na população latina.
a cura do sofrimento dependerá então da eliminação de todas as células malignas existentes no paciente. Em geral, algumas das leucemias podem ser suscetíveis à cura apenas com quimioterapia, mas hoje em dia deve-se dar muita importância aos chamados fatores prognósticos que se baseiam em modelos matemáticos que permitem localizar os pacientes no grau de prognóstico que têm e incluem:
1. O tipo de leucemia.
2. Alteração molecular inicial e sua persistência apesar do tratamento ou erradicação.
3. A Idade. Pacientes com mais de 60 anos têm um prognóstico ruim em comparação com pacientes menores de idade.
4. Quimioterapia. Devem ser utilizados os medicamentos indicados e, sobretudo, as doses recomendadas. Por exemplo na LAL do adulto, o uso do esquema HyperCyVAD (doses escaladas de ciclofosfamida, vincristina, adriamicina, dexametasona em combinação com arabinosido de citosina e methotrexate) alcança 90% de RC e cura em 50% dos casos, dados não antes vistos com outros esquemas. Este tratamento é tóxico e requer que seu uso seja apenas em instituições que tenham recursos de apoio suficientes. Infelizmente, em nosso meio nem todos os centros contam com os medicamentos recomendados e os resultados não serão reprodutíveis, já que também não contam com suficiente apoio para o paciente durante a fase de máxima mielossupressão.
5. Terapia de apoio. As conquistas da quimioterapia e novos medicamentos com novas combinações e com mais especificidade obrigam a implementação de grupos multidisciplinares com a direção do hematologista; instalação e uso de cateteres centrais; suporte de banco de sangue para o Suporte de transfusões de plaquetas e eritrócitos (inclusive produtos radiados); a intervenção do Infectologista para a detecção de infecções e o uso adequado de antibióticos ou antifúngicos, sejam profiláticos ou terapêuticos, se; a habilitação de salas de isolamento com serviços de manutenção e Intendência que permitam alcançar um ambiente livre de bactérias, inclusive a alimentação estéril; um laboratório com protocolo de manejo de amostras de pacientes em protocolos de tratamento específico e o manejo de amostras especiais (preparação de concentrados de leucócitos na citometria hemática buffy coat e conseguir uma leitura ótima); área de preparação de medicamentos de alta especialização, e, por outro lado, o mais importante é o pessoal de enfermagem, auxiliar e administrativo, que com todo o grupo se esperam os resultados que têm em outros países.
6. Transplante de medula óssea (MO). É um tipo de tratamento complexo e de alto custo, que requer de um doador de MO compatível e de um padecimento inativo com alta probabilidade de recaída precoce ou tardia ou, com fatores de mau prognóstico. É um procedimento com tendências mais curativas, pois utiliza megadoses de quimioterapia para erradicar as células leucêmicas, mas na tentativa, também erradica os precursores normais e torna-se necessária a reposição de uma nova medula normal compatível. O tipo alogênico (irmão compatível idêntico) é a melhor seleção já que o receptor o Aceita apenas por identidade parcial (os únicos de aceitação total são os gêmeos idênticos) e portanto produz uma rejeição do enxerto contra o hospedeiro, o que por sua vez produz enxerto contra leucemia e aumenta a erradicação da clona leucêmica, porém, em casos graves da síndrome (graus 3-4) a morbimortalidade aumenta apesar do manejo específico.
LEUCEMIAS crônicas
1. Leucemia linfocítica crónica. Ocorre mais frequentemente em pessoas mais velhas e o critério é a persistência de linfocitose de mais de 10 x 109/l, E MO com infiltração de mais de 50% de linfócitos com fenótipo CD5+. O critério de administrar tratamento é a duplicação da contagem de linfócitos em um ano ou a progressão de adenomegalia ou esplenomegalia, embora alguns casos escapem deste critério pela presença de anemia hemolítica ou trombocitopenia autoimune e então se indica o tratamento à base da combinação de fludarabina, ciclofosfamida e prednisona, nos estágios I e II só somenter a observação e esperar evolução sem tratamento.
2. Leucemia mielóide crónica (LMC). Nesta doença existe um grande avanço no conhecimento da presença do cromossomo Filadélfia, que foi descrito em 1950 nessa cidade da União Americana e que em seus inícios significou o primeiro marcador cromossômico em associação à malignidade, porém ao passar dos anos de pesquisa, conseguiu-se conhecer a t(9;22) com a expressão funcional do cromossoma com a produção de uma oncoproteína com grande actividade de tirocinocinase, que aumenta a proliferação celular e que por sua vez explica a grande leucocitose e trombocitose com que se apresentam estes pacientes, assim como a grande esplenomegalia (figura 5).
por estas datas são cumpridos de 10 a 12 anos da descoberta de uma pequena molécula dirigida especificamente contra esse substrato molecular doador de fosfatos para a regulação interna da célula leucêmica e seus substratos. O inicialmente chamado STI (signal transduction inibitor) produziu inibição competitiva da fosforilação, levou a célula à apoptose e os pacientes alcançaram resultados clínicos nunca antes vistos com remissões moleculares de até 80-90% a 10 anos, o que mudou dramaticamente a história natural da doença, que com o pré-tratamento não era superior a 3 anos. Esta informação faz uma mudança na história natural e prognóstico da doença, antes mortal a curto prazo.

estes exemplos são uma base do avanço tão intenso que ocorreu nos últimos anos e que é desejável que permita provocar o interesse dos estudantes de medicina, dos seus professores, mas sobretudo das autoridades educativas de que a leucemia em geral ocupa os primeiros 5 lugares de frequência das doenças malignas do adulto e dos primeiros lugares nas crianças, pelo que a hematologia deverá incluir-se dentro das matérias básicas no curriculum de a carreira de medicina e ainda como parte dos cursos de especialização de pós-graduação.
embora seja útil por sua simplicidade, a classificação franco-americana britânica (FAB) pode levar a erros diagnósticos e, portanto, terapêuticos em até 20% dos casos. Por este motivo, a classificação por métodos de imuno-histoquímica e biologia molecular tornou-se um requisito sine qua non para a correta classificação e posterior manejo dos pacientes (figura 6).

ao contrário da classificação FAB, a da OMS (Tabela 2) reflete uma mudança no paradigma através do qual entendemos as doenças hemáticas, pois pela primeira vez se uniram a informação genética, as características morfológicas, citoquímicas e imunofenotípicas com os achados clínicos dentro dos algoritmos diagnósticos das neoplasias do tecido hematopoiético; a importância relativa de cada critério difere entre neoplasias e não existe um “padrão ouro” para a classificação de todas as doenças hematológicas malignas. O objetivo foi definir entidades que pudessem ser reconhecidas pelos patologistas e que tivessem relevância clínica. Desde a sua aparição em 2001 foram feitas diferentes revisões para atualizar seu conteúdo em relação às descobertas mais atuais. A última revisão é de 2008 e classifica as neoplasias malignas hematológicas da seguinte forma:
1. Neoplasias mielóides.
2. Neoplasias linfóides.
3. Doenças de mastócitos ou primers.
4. Doenças histiocíticas e de células dendríticas.

neoplasia mielóide
as neoplasias mielóides são derivadas de progenitores na medula óssea, que diferem em eritrócitos, granulócitos (neutrófilos, basófilos e eosinófilos), monócitos e megacariócitos. Na classificação FAB são reconhecidas 3 categorias principais:
1. Leucemia mielóide aguda.
2. Síndromes mielodisplásicas.
3. Neoplasias mieloproliferativas.
os determinantes mais importantes das categorias reconhecidas utilizam suas características morfológicas, histoquímicas e imunofenotípicas e são a porcentagem de blastos, a linhagem celular e o grau de diferenciação das células neoplásicas (figura 7). Nos últimos anos, as características genéticas (citogenéticas e moleculares), bem como o tratamento prévio e a evolução da mielodisplasia, mostraram impacto significativo no comportamento clínico desses padecimentos que nem sempre se correlacionam adequadamente com as categorias da FAB, pelo que o debate central para sua reclassificação foi o discriminar entre as entidades patológicas e os fatores prognósticos, para conseguir uma classificação com relevância clínica e significância para o patologista. Algumas anormalidades genéticas parecem definir diferentes doenças, enquanto outras representam fatores prognósticos de uma doença específica.

atualmente, a classificação da OMS reúne os padecimentos mielóides em 4 grupos principais:
1. Doenças mieloproliferativas
2. Síndromes mielodisplásicas
3. Doenças mielodisplásicas / mieloproliferativas
4. Leucemias agudas mielóides

as doenças mieloproliferativas são um grupo de distúrbios clonais associados à proliferação de uma ou mais linhas mielóides. Torna-se cada vez mais claro que estas doenças são frequentemente associadas a mutações que resultam em aumentos anormais da actividade das tirosino-quinases e na proliferação de células progenitoras da medula óssea, independentes de factores de crescimento. A porcentagem de blastos na medula óssea é normal ou ligeiramente elevada, mas é sempre inferior a 20%. A hematopoiese é geralmente eficaz, resultando em um aumento nas contas de uma ou mais células maduras no sangue periférico. O protótipo das neoplasias mieloproliferativas é a leucemia mielóide crônica cromossomo Filadélfia positivo (Ph1) (BCR/ABL). As outras entidades incluídas são:
1. Policitemia vera.
2. Mielofibrose idiopática.
3. Trombocitemia essencial primária.
4. Leucemia eosinofílica crónica.
5. Leucemia neutrofílica crónica.
6. Mastocitose.
7. Neoplasias mieloproliferativas Não Classificáveis.

síndromes mielodisplásicas referem-se a distúrbios caracterizados por produção celular ineficaz e displasia, com risco variável de transformação em leucemia aguda. A celularidade na medula é muitas vezes aumentada, mas é muito variável. Há maturação, mas também displasia de uma ou mais linhas mielóides. A hematopoiese não é eficaz e, portanto, existem citopenias. Esta rubrica inclui:
1. Citopenia refratária com displasia* de uma linha.
• Anemia refractária.
• Neutropenia refractária.
• Trombocitopenia refractária.
2. Anemia refractária com sideroblastos anulares.
3. Citopenia refratária com displasia de múltiplas linhagens.
4. Anemia refractária com excesso de blastos.
5. Síndrome mielodisplásica com d (5Q).
6. Síndrome mielodispásica inclassificável.
7. Síndrome mielodisplásica juvenil, inclui uma entidade provisória conhecida como citopenia refractária juvenil.
as síndromes mielodisplásicas / mieloproliferativas incluem distúrbios nos quais coexistem características displásicas e proliferativas. Neste grupo iclui-se a leucemia juvenil mielomonocítica, a qual é representativa de ambas as síndromes (mielodisplásico e mieloproliferativo). Quase metade dos pacientes apresenta-se com contas de neutrófilos normais ou baixas e displasia de múltiplas linhas celulares sem organomegalia e medula óssea com morfologia que semeia a anemia refratária com excesso de blastos, mas com monocitose. Outros doentes apresentam neutrofilia intensa, monocitose e esplenomegalia. Ainda não se sabe se são 2 doenças diferentes, uma mielodisplásica e uma mieloproliferativa; no entanto, até ao momento, não existem diferenças nas anomalias citogenéticas nem nos padrões de crescimento das Colónias in vitro ou na sua evolução clínica, pelo que existe controvérsia entre clínicos e patologistas de acordo com o seu lugar dentro da classificação. De acordo com a última revisão, Nesta categoria estão localizados:
1. Leucemia mielomonocítica crónica.
2. Leucemia mielóide crônica atípica (negativa para BCR / ABL).
3. Leucemia mielomonocítica juvenil.
4. Síndrome mielodisplásica / mieloproliferativa não classificável.
na categoria das leucemias agudas mieloblásticas (LAM) (a qual é definida por uma percentagem superior a 20% de mieloblastos na medula óssea ou em sagre periférica, ou a presença de uma anormalidade citogenética em particular apesar da contagem de blastos) é reconhecida aos seguintes grupos:
1. LAM com translocações citogenéticas recorrentes.
2. LAM com características mielodisplásicas.
3. LAM e SMD relacionados a tratamentos antineoplásicos.
4. LAM não classificável.
5. Sarcoma mielóide.
6. Proliferações mielóides relacionadas à síndrome de Down.
7. Neoplasia blástica plasmocitóide de células dendríticas.
NEOPLASIAS linfóides
são aquelas que se originam das células que normalmente se desenvolvem em linfócitos T (lt citotóxicos, colaboradores ou reguladores) ou linfócitos B (linfócitos ou células plasmáticas). Em geral, as neoplasias linfoides se dividem naquelas que derivam de precursores linfoides e aquelas provenientes de linfócitos maduros e células plasmáticas e posteriormente se agrupam de acordo com sua estirpe (B ou T).
historicamente, as neoplasias linfóides que se apresentam na medula óssea e que envolvem a medula óssea foram separadas daquelas que se apresentam como tumor (linfoma). No entanto, sabe-se agora que qualquer linfoma pode apresentar características clínicas de leucemia e que qualquer leucemia pode ocasionalmente se apresentar como um tumor (sarcoma granulocítico). Na classificação da OMS, o diagnóstico de várias neoplasias linfóides depende não apenas da localização anatômica das células tumorais, mas da origem da célula tumoral morfologicamente definida. Tais considerações anularam a relevância dos Termos L1 e L2 da classificação FAB, pois não correlacionam com seu imunofenótipo, anormalidades genéticas ou com seu curso clínico (figura 8). A L3 é equivalente ao linfoma de Burkitt em fase leucêmica e deve ser diagnosticada como tal.

1. Neoplasias de precursores. Existe consenso de que as neoplasias de precursores que se apresentam como tumores sólidos e aqueles que envolvem medula óssea e sangue são biologicamente a mesma doença com diferentes apresentações clínicas. A maioria das neoplasias de precursores linfóides é apresentada como leucemias, pelo que foi acordado que a classificação deve manter o termo LAL para a fase leucêmica das neoplasias precursoras de tipos B E T. existem 2 categorias principais:
* Leucemias / linfomas precursoras B.
• Leucemias/linfomas precursores t.
2. Neoplasias de células B maduras. A classificação proposta considera os linfomas e leucemias do mesmo tipo celular como uma mesma doença com diferentes apresentações clínicas ou estádios. As doenças específicas derivadas das células B maduras são as seguintes:
1. Leucemia linfocítica crônica / linfoma de pequenos linfócitos.
2. Linfoma linfoplasmacítico.
3. Linfoma de células do manto.
4. Leucemia prolinfocítica das células B.
5. Linfoma folicular.
6. Linfoma difuso de células grandes b.
• Linfoma intravascular de células grandes b.
• Linfoma mediastinal primário de células grandes B.
• Linfoma de células grandes B (relacionado ao vírus Epstein Barr-VEB).
• Linfoma de células grandes B rico em histiócitos e células T.
• Linfoma difuso de células grandes B do sistema nervoso central.
• Linfoma difuso de células grandes B cutâneo primário.
• Linfoma difuso de células grandes B do idoso positivo para VEB.
• Linfoma pasmablástico.
• Linfoma pleural primário.
• Linfoma de células grandes B positivo para alkoma (ALK).
• Linfoma de Burkitt.
7. Linfoma de células B da zona marginal.
8. Linfoma de células B da zona marginal extranodal.
9. Linfoma de células B da zona marginal esplénica
10. Leucemia de células peludas.
11. Plasmocitoma / mieloma das células plasmáticas.

NEOPLASIAS de linhas mielóides e linfóides
algumas neoplasias expressam marcadores de linhas mielóides e linfóides, este grupo representa as leucemias de linhagem ambígua, que são aquelas que ou não apresentam características de linha linfóide ou mielóide (leucemia aguda indiferenciada) ou apresentam características de ambas as linhas (leucemia aguda de fenótipo misto ou de linhas mistas).
Tabela 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍABassan R, Hoelzer D. Modern Therapy of Acute Lymphoblastic Leukemia. J Clin Oncol. 2011;29:523-43. Burnett a, Wetzler M, Löwenberg B. Therapeutic advances in Acute Leukemia. J Clin Oncol. 2011;29:487-94. Campo e, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasmas and beyond: developing concepts and practical applications. Sangue. 2011 May 12; 117(19):5019-32. Epub 2011 Fev 7. Revisao.
Cortes J, Hochhaus a, Hugues T, Kantajian H. linha de frente e terapias de salvamento com inibidores da tirosina cinase e outros tratamentos na leucemia mielóide crónica. J Clin Oncol. 2011;29:524-31. Chin-Hon Pui, Carroll WL, et al. Biology, Risk Stratification, and Therapy of Pediatric Acute Leukemia: An Update. J Clin Oncol. 2011;29:551-65. Gambacorti PC, Antolin L, Hurtado Mr. Journal National Cancer Institute. 2011;103:1-9. Grever M, Lozanki G. Estratégias modernas para leucemia de células cabeludas. J Clin Oncol. 2011:29;583-90.
Gribben JG, O’Brien S. Update on Therapy of Chronic Lymphocytic Leukemia. J Clin Oncol. 2011;29:544-50. Hurtado Mr, Vargas VP, Cortes FJ. Leucemia Mielóide Crónica. Conceitos atuais em fisiopatologia e tratamento. Cancelología. 2007:2:137-47. Hurtado Mr, Vargas VP, et al. O Imatinib foi comparado com Imatinib/citarabina no tratamento de primeira linha da leucemia mielóide crónica positiva para o cromossoma Filadélfia precoce. Resultados de um ensaio clínico aleatorizado do grupo de leucemia colaborativa mexicana. Leucemia Clínica. 2008: 2(2);1128-32. Lichtman MA. Classificação e manifestações clínicas das doenças mielóides clonais. En: Williams. Hematologia. Mc Graw-Hill; 2010. Marcucci G, Haferlach T, Dohner H. Molecular Genetics of adult acute mieloid Leukemia. Implicações prognósticas e terapêuticas. J Clin Oncol. 2011;29:475-86. Rafael Hurtado m Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rev Fac Med UNAM. 2010; 53:36-43.
Sanz M, Lo-Coco F. Modern Approaches to treating Acute Promyelocytic Leukemia. J Clin Oncol. 2011;29:495-503.
Nota
* Displasia. Refere-se à alteração citomorfológica que inclui dissociação da maturação núcleo-citoplasma (lembre-se que a maturação da cromatina depende do DNA e do citoplasma do RNA, portanto o núcleo pára na maturação enquanto o citoplasma segue seu processo normal) o que produz células não viáveis e há apoptose intramedular.