overzichtsartikel
leukemie-voor de huisarts
leukemie voor de huisarts
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
hoofd van de afdeling Hematologie. Hospital Ángeles del Pedregal. Mexico, DF. E-mailadres: [email protected]
B intern geneesmiddel. Hospital Ángeles del Pedregal. Mexico. DF.
c intern geneesmiddel. Hospital Ángeles del Pedregal. Mexico. DF.
ontvangen: 17 oktober 2011
geaccepteerd: 07 januari 2012
inleiding
ondanks de grote moleculaire en therapeutische vooruitgang in de studie van leukemias, zijn de fundamentele aspecten van deze aandoening nog niet duidelijk bekend bij de niet-hematoloog, dus het doel van dit werk is om fundamentele informatie te verstrekken aan medische studenten en artsen in het algemeen, en dat maakt het vooral mogelijk om algemene kennis van leukemias te verkrijgen, hun tijdige diagnose en vroeg te raadplegen met de hematoloog.

definitie
leukemie is de term die wordt gebruikt om een groep maligne bloedziekten te definiëren. Vroege diagnose is essentieel, omdat het de patiënt in staat zal stellen om vroeg te gaan met de hematologiespecialist, die het diagnostische proces zal leiden en de specifieke behandeling zal aanbieden. Het wordt gekenmerkt door het hebben van een klonale, autonome en abnormale proliferatie van de cellen die aanleiding geven tot de rest van de normale cellen van het bloed (tumorgedrag in het algemeen).
Dit impliceert dat een vroege cel een genetische verandering ondergaat die een abnormale kloon (kolonie) van zichzelf zal veroorzaken om ongecontroleerd te voorkomen. Deze abnormale productie is verstoord omdat de abnormale cellen vermenigvuldigen in Beeld en gelijkenis van zichzelf, zodat ze geleidelijk bezetten de ruimte van het normale beenmerg en leiden tot progressieve bloedarmoede, abnormale bloeden en aanleg voor infecties. Aan de andere kant, wanneer abnormale cellen binnenvallen andere weefsels, zal er falen van de werking van het betrokken orgaan, bijvoorbeeld infiltratie naar het centrale zenuwstelsel dat optreedt in acute lymfoblastische leukemie (LAL) kan manifesteren met hoofdpijn, epileptische aanvallen, gerichte motorische veranderingen, verhoogde intracraniale druk, en het niet maken van een vroege diagnose en een adequate behandeling, zal het verlies van functie en onomkeerbare gevolgen presenteren.
klinische manifestaties
het klinische beeld is divers en hangt af van het type leukemie: acuut of chronisch, maar voor de 2 zijn er niet-specifieke klinische manifestaties (die bij elke ziekte voorkomen):
1. Vermoeidheid.
2. Gemakkelijke vermoeidheid.
3. Algemene zwakte.
4. Wil rusten of in bed blijven.
5. Het vereist de hulp van iemand om aan uw persoonlijke behoeften te voldoen.
chronische leukemieën zijn indolent en tot 50% van de gevallen worden ontdekt in een routine klinisch onderzoek of laboratoriumonderzoek bij vrijwilligers die als gezond worden beschouwd en bloed komen doneren.

in acute vormen worden specifieke manifestaties afgeleid van deficiëntie van een van de cellijnen:
1. Erytrocyten: bloedarmoede syndroom waarvan de intensiteit zal afhangen van de mate van hypoxemie, ongeacht de mate van bloedarmoede. Dyspneu van middelmatige inspanning tot orthoprea.
2. Bloedplaatjes: petechiën, ecchymose in extremiteiten en in ernstigere gegeneraliseerde gevallen, droge en natte bloeding met epistaxis, gingivorrhagia, hematurie, manen of hematochesie. Zeer ernstig in het centrale zenuwstelsel (CZS).
3. Leukocyten: koorts, diaforese, gelokaliseerde infecties tot frank septicemia (bacteriën of schimmels). Ze komen voor met neutropenie van minder dan 250 neutrofielen in totaal/mm3.
infiltratief syndroom: verwijst naar abnormale implantatie in elk weefsel, hoewel het vaak voorkomt:
1. Hepatomegalie of splenomegalie (Figuur 5).
2. Adenomegalie (lokaal of gegeneraliseerd).
3. Leukemische teint.
4. Botpijn door beenmerguitzetting.
5. Zachte weefsels (granulocyt sarcoom).
6. Testikel.
7. SNC.
8. Tandvlees en elke plaats (figuur 1).

metabole stoornissen: ze zijn het gevolg van abnormale hyperproductie van kwaadaardige cellen en verhoogde apoptose.
1. Acidose.
2. Verhoogd lactaatdehydrogenase (LLD).
3. Hyperkaliëmie.
4. Hyperurikemie.
5. Verhoogd β2 microglobuline.

Het klinische bewijs overheerst als de hoeksteen van de vermoedelijke diagnose van acute leukemieën en van enige voorwaarde, maar wat volgt is een aanvulling op de diagnose met de ondersteuning van het klinisch laboratorium in de cytometrie volledig bloedbeeld, of speciale, dat is te zeggen, de grondige observatie van het perifere bloed uitstrijkje door de technische medewerkers die de voorbereiding in de identificatie van abnormale cellen, met name leukemie.

Laboratoriumwijzigingen die een speciale beoordeling vereisen zijn onder meer:
1. Bloedarmoede (elke graad).
2. Leukopenie of leukocytose (overheersing van een cellijn).
3. Trombocytopenie.
4. Combinaties: bicitopenie of pancytopenie.
extra voorzichtigheid is geboden wanneer het laboratorium de aanwezigheid van leukocyten of atypische lymfocyten meldt (kunnen leukemische blasten zijn). Het is raadzaam om een deskundigenonderzoek te vragen (Figuur 2).

beenmerg aspiratie is essentieel voor de diagnose (Figuur 3) en 20% van de blasten zijn nodig om de criteria voor acute leukemie in een van de variëteiten vast te stellen.

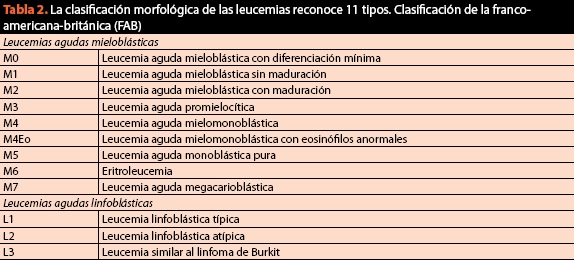
In dezelfde procedure moeten monsters worden genomen voor de definitieve classificatie van de aandoening en moeten karyotype en immunofenotype worden aangevraagd, aangezien het cytomorfologische criterium momenteel van vitaal belang is, maar niet langer volstaat. De huidige en huidige classificatie van kwaadaardige bloedziekten wordt weergegeven in Tabel 2.
de behandeling is gericht op twee belangrijke aspecten: de eerste is het specifieke anti-lekemisch en is gebaseerd op het gebruik van geneesmiddelen van chemische oorsprong die bekend staan als chemotherapie, waarvan het hoofddoel is om alle leukemische cellen uit het lichaam uit te roeien. Het tweede aspect van de behandeling is ondersteuning voor complicaties die meestal optreden bij patiënten bij opname.
de behandeling is gericht op twee belangrijke aspecten: de eerste is de specifieke antileukemie en is gebaseerd op het gebruik van geneesmiddelen van chemische oorsprong die bekend staan als chemotherapie, waarvan het hoofddoel is om uit te roeien, dat wil zeggen alle leukemische cellen uit het lichaam te verwijderen.
het tweede aspect van de behandeling is de ondersteuning van de complicaties die patiënten gewoonlijk vertonen bij opname, zoals:
1. Bloedarmoede.
2. Abnormale bloeding.
3. Pulmonale en gegeneraliseerde infecties, onder andere (figuur 4).
4. Om het even welke andere aangrenzende complicaties die de patiënt kan hebben (comorbiditeit), zoals reeds bestaande voorwaarden, b.v. diabetes, hypertensie, hartkwaal, en andere ziekten gemeenschappelijk onder patiënten die aan leukemie lijden.


daarom is het zeer belangrijk om er rekening mee te houden dat de behandeling tegen leukemie multidisciplinair is, waarbij ook andere specialisten betrokken zijn, zoals ondersteuning van de hematoloog.
Antileukemische behandeling zal ook verschillend zijn voor verschillende soorten leukemie en voor acute vormen. Het is verdeeld in 3 fasen:
1. Inductie van remissie. Het doel is om volledige remissie (CR) te bereiken, dat wil zeggen, normalisatie van de bloedwaarden van de patiënt, de afwezigheid van symptomen of tekenen dat leukemie aanhoudt met infiltratie. Tijdens het proces moet de patiënt een “leukemie-vrije toestand” in het beenmerg hebben en de toekomst moet het herstel zijn naar een normale hematopoiese, en helaas in andere gevallen herstellen ze met de ziekte, die spreekt van resistente leukemie waarvan de prognose verschrikkelijk is. Dit eerste proces kan 6 tot 8 weken duren om CR te bereiken.
2. Consolidatie. Het gaat om het gebruik van dezelfde geneesmiddelen die werden gebruikt bij de inductie of combinatie van andere chemotherapeutica, ook met het oog op het volgen van de uitroeiing van resterende kwaadaardige cellen die resistentie tegen eerste gebruik kunnen ontwikkelen.
3. Onderhoud. Het verdient de voorkeur om de patiënt onder het effect van chemotherapie te houden vóór de mogelijkheid van beginnende leukemische activiteit en dat met de behandeling het effect behoudt totdat de ziekte verdwijnt.

tot nu toe worden voor acute vormen de strengste criteria om CR als complexer te beschouwen, aangezien het het beste is om te zoeken naar moleculaire remissie, waarbij wordt gezocht naar de initiële chromosomale verandering zoals in het geval van promyelocytaire leukemie (M3-FAB) met t (15).;17) aanvankelijk moet worden gezocht naar specifieke moleculaire en cytogenetische studies ondanks het feit dat in CR, omdat als de translocatie aanhoudt, dan de agressieve behandeling moet worden voortgezet tot de totale eliminatie van de kloon. Deze verscheidenheid van leukemie zou als potentieel geneesbaar moeten worden beschouwd en is één van gemeenschappelijkste in de latinobevolking.
de genezing van de ziekte zal dan afhangen van de eliminatie van alle bestaande kwaadaardige cellen in de patiënt. In het algemeen kunnen sommige leukemieën vatbaar zijn voor genezing met chemotherapie alleen, maar vandaag de dag moet veel belang worden gehecht aan zogenaamde prognostische factoren die gebaseerd zijn op wiskundige modellen die het mogelijk maken patiënten te plaatsen in de mate van prognose die ze hebben en omvatten:
1. Het type leukemie.
2. De initiële moleculaire verandering en de persistentie ervan ondanks behandeling of uitroeiing.
3. leeftijd. Patiënten ouder dan 60 jaar hebben een slechte prognose in vergelijking met jongere patiënten.
4. Chemotherapie. De vermelde geneesmiddelen en vooral de aanbevolen doses dienen te worden gebruikt. Bij volwassen LAL bijvoorbeeld bereikt het gebruik van het HyperCyVAD-schema (verhoogde doses cyclofosfamide, vincristine, adriamycine, Dexamethason in combinatie met cytosine arabinoside en methotrexaat) 90% CR en geneest in 50% van de gevallen, gegevens die niet eerder werden gezien met andere schema ‘ s. Deze behandeling is giftig en vereist dat het alleen wordt gebruikt in instellingen die over voldoende ondersteunende middelen beschikken. Helaas, in onze omgeving niet alle centra hebben de aanbevolen medicijnen en de resultaten zullen niet reproduceerbaar zijn, omdat ze niet genoeg ondersteuning voor de patiënt tijdens de fase van maximale myelosuppressie.
5. Ondersteunende therapie. De resultaten van de chemotherapie en nieuwe geneesmiddelen, nieuwe combinaties en met meer specificiteit dwingen de implementatie van de multidisciplinaire teams met het adres van de hematoloog; installatie en gebruik van centrale veneuze katheters; steun voor bloedbank voor de ondersteuning van transfusies van bloedplaatjes en erytrocyten (zelfs producten stellate); de interventie van de infectoloog voor de detectie van infecties, en het juiste gebruik van antibiotica of antischimmelmiddelen, hetzij profylactische of therapeutische, indien nodig; het ter beschikking stellen van isolatiekamers met onderhoud en kwartiermeester diensten die het mogelijk maken om een bacterievrije omgeving te bereiken, inclusief steriele voeding; een laboratorium met patiëntmonsterbeheerprotocol in specifieke behandelprotocollen en het beheer van speciale monsters (bereiding van leukocytenconcentraten in buffy coat hematic cytometry en het bereiken van een optimale aflezing); gebied van voorbereiding van zeer gespecialiseerde geneesmiddelen, en aan de andere kant, de belangrijkste is het verplegend, hulp-en administratief personeel, die met de hele groep worden verwacht de resultaten die ze hebben in andere landen.
6. Beenmergtransplantatie (MO). Het is een complexe en dure behandeling die een compatibele MO-donor en een inactieve aandoening met een hoge kans op een vroege of late terugval of, met factoren van slechte prognose vereist. Het is een procedure met meer curatieve neigingen omdat het megadoses chemotherapie gebruikt om de leukemische cellen uit te roeien, maar in de poging, het ook de normale voorlopers uitroeit en de vervanging van een nieuw compatibel normaal merg is noodzakelijk. Het type allogene (broer of zus compatibel identiek) is de beste selectie aangezien de ontvanger slechts door gedeeltelijke identiteit aanvaardt (de enige van totale aanvaarding is identieke tweelingen), en daarom resulteert in een afwijzing van de graft-versus-host ziekte, die op zijn beurt tot graft-versus-leukemie leidt en de uitroeiing van de leukemische klonen verhoogt, echter, in ernstige gevallen van het syndroom (graad 3-4) wordt de morbiditeit en mortaliteit verhoogd ondanks de specifieke behandeling.
chronische leukemieën
1. Chronische lymfatische leukemie. Het komt vaker voor bij oudere mensen en het criterium is de persistentie van lymfocytose van meer dan 10 x 109 / l, en MO met infiltratie van meer dan 50% van lymfocyten met CD5+ – fenotype. Het criterium van de behandeling is de duplicatie van de rekening van lymfocyten in een jaar of progressie van adenomegalie of splenomegalie, hoewel sommige gevallen voorbij deze standaard door de aanwezigheid van hemolytische anemie of trombocytopenie, auto-immuun, en vervolgens aangegeven de behandeling op basis van de combinatie van fludarabine, cyclofosfamide en prednison, in de stadia I en II alleen somenter en waargenomen en verwachte evolutie zonder behandeling.
2. Chronische myeloïde leukemie (CML). In deze ziekte is er een grote vooruitgang in de kennis van de aanwezigheid van het Philadelphia-chromosoom, dat in 1950 in de stad van de Amerikaanse Unie werd beschreven, en dat de oprichting ervan het eerste markerchromosoom in samenwerking met maligniteit was, maar door de jaren van onderzoek, slaagden wij erin om t(9;22) met de functionele expressie van het chromosoom met de productie van een oncoproteïne met grote thyrokinokinase activiteit, die celproliferatie verhoogt en die op zijn beurt de grote leukocytose en trombocytose verklaart waarmee deze patiënten aanwezig zijn, evenals de grote splenomegalie (Figuur 5).
op dit moment zijn er 10 tot 12 jaar verstreken sinds de ontdekking van een klein molecuul dat specifiek gericht is tegen dit fosfaatdonor moleculair substraat voor de interne regulatie van de leukemische cel en zijn substraten. De aanvankelijk genoemd STI (signal transduction inhibitor) produceerde competitieve remming van de fosforylering, leidde tot celapoptose en de patiënten bereikten klinische resultaten nooit eerder gezien met moleculaire remissies van maximaal 80-90% bij 10 jaar, die dramatisch de natuurlijke geschiedenis van de ziekte veranderde, met de voorbehandeling was niet meer dan 3 jaar. Deze informatie maakt een verandering in de natuurlijke geschiedenis en prognose van de ziekte, eerder fataal op de korte termijn.

Deze voorbeelden zijn een basis van de vooruitgang die de laatste jaren zo intens is opgetreden en dat het wenselijk is toe te staan dat de belangstelling van studenten geneeskunde, hun leraren, maar vooral van de onderwijsautoriteiten wordt gewekt voor leukemie in het algemeen, die de eerste 5 plaatsen inneemt van de frequentie van kwaadaardige ziekten bij volwassenen en van de eerste plaatsen bij kinderen, zodat de hematologie wordt opgenomen in de kernvakken van het curriculum van de loopbaan van de geneeskunde en zelfs als onderdeel van de cursussen postdoctorale specialisatie.
hoewel nuttig voor zijn eenvoud, kan de British Franco-American classification (FAB) leiden tot diagnostische en dus therapeutische fouten in maximaal 20% van de gevallen. Om deze reden is classificatie door immunohistochemie en moleculaire biologie methoden een conditio sine qua non geworden voor de juiste classificatie en daaropvolgende behandeling van patiënten (Figuur 6).

in tegenstelling tot de classificatie FAB, weerspiegelt de WHO (tabel 2) een verandering in het paradigma waardoor we bloedziekten begrijpen, omdat voor het eerst de genetische informatie, de morfologische, cytochemische en immunofenotypische gegevens werden gecombineerd met de klinische bevindingen binnen de diagnostische algoritmen van neoplasmata van het hematopoëtische Weefsel.; het relatieve belang van elk criterium verschilt tussen neoplasmata en er is geen “gouden standaard” voor de classificatie van alle hematologische maligniteiten. Het doel was om entiteiten te definiëren die door pathologen konden worden herkend en die klinische relevantie hadden. Sinds de verschijning in 2001 zijn verschillende herzieningen gemaakt om de inhoud ervan te actualiseren in relatie tot de meest recente ontdekkingen. De laatste beoordeling was uit 2008 en classificeert hematologische maligniteiten als volgt:
1. Myeloïde neoplasmata.
2. Lymfoïde neoplasmata.
3. Mestcel-of vetcelziekten.
4. Histiocytische en dendritische celziekten.

neoplasis myeloïde
myeloïde neoplasmata zijn afgeleid van voorlopercellen in het beenmerg, differentiëren zich tot erytrocyten, granulocyten (neutrofielen, basofielen en eosinofielen), monocyten en megakaryocyten. De FAB-classificatie herkent 3 hoofdcategorieën:
1. Acute myeloïde leukemie.
2. Myelodysplastische syndromen.
3. Myeloproliferatieve neoplasmata.
de belangrijkste determinanten van de met behulp van morfologische, histochemische en immunofenotypische categorieën erkende categorieën en het percentage blastcellen, de cellijn en de mate van differentiatie van de neoplastische cellen (figuur 7). In de afgelopen jaren toonden de genetische kenmerken (cytogenetische en moleculaire), evenals de voorbehandeling en de evolutie van myelodysplasie, een significante invloed op het klinische gedrag van dit lijden, die niet altijd goed correleren met de categorieën van de FAB, zodat het centrale debat voor herclassificatie was om onderscheid te maken tussen de entiteiten en pathologische prognostische factoren, om een classificatie met klinische relevantie en betekenis voor de patholoog te bereiken. Sommige genetische afwijkingen lijken verschillende ziekten te definiëren, terwijl andere prognostische factoren voor een specifieke ziekte vertegenwoordigen.

momenteel groepeert de WHO-classificatie myeloïde aandoeningen in 4 hoofdgroepen:
1. Myeloproliferatieve ziekten
2. Myelodysplastische syndromen
3. Myelodysplastische / myeloproliferatieve ziekten
4. Acute myeloïde leukemieën

myeloproliferatieve ziekten zijn een groep klonale stoornissen geassocieerd met de proliferatie van één of meer myeloïde lijnen. Het wordt steeds duidelijker dat deze ziekten vaak geassocieerd worden met mutaties die een abnormale toename van de tyrosinekinaseactiviteit en groeifactor-onafhankelijke progenitorcelproliferatie in het beenmerg veroorzaken. Het percentage blasten in het beenmerg is normaal of licht hoog, maar altijd minder dan 20%. Hematopoiese is meestal effectief, wat resulteert in een toename van de tellingen van een of meer rijpe cellen in perifeer bloed. Het prototype van myeloproliferatieve neoplasmata is Philadelphia chromosoom positieve (Ph1) chronische myeloïde leukemie (BCR/ABL). De andere opgenomen entiteiten zijn:
1. Polycythemia vera.
2. Idiopathische myelofibrose.
3. Primaire essentiële trombocytemie.
4. Chronische eosinofiele leukemie.
5. Chronische neutrofiele leukemie.
6. Mastocytose.
7. Niet-classificeerbare myeloproliferatieve neoplasmata.

myelodysplastische syndromen verwijzen naar stoornissen gekenmerkt door ineffectieve celproductie en dysplasie, met een variabel risico op transformatie naar acute leukemie. Cellulariteit in het merg is vaak verhoogd, maar zeer variabel. Er is rijping maar ook dysplasie van een of meer myeloïde lijnen. Hematopoëse is niet effectief en daarom bestaan er cytopenieën. Deze post omvat:
1. Refractaire cytopenie met één-lijn dysplasie*.
• refractaire anemie.
• refractaire neutropenie.
• refractaire trombocytopenie.
2. Refractaire bloedarmoede met ringvormige sideroblasten.
3. Refractaire cytopenie met dysplasie van meerdere geslachten.
4. Refractaire bloedarmoede met overmatige ontploffing.
5. Myelodysplastisch syndroom met d (5q).
6. Onclassificeerbaar myelodyspastisch syndroom.
7. Juveniele myelodysplastisch syndroom, omvat een voorlopige entiteit die bekend staat als juveniele refractaire cytopenie.
myelodysplastische / myeloproliferatieve syndromen omvatten aandoeningen waarbij dysplastische en proliferatieve kenmerken naast elkaar bestaan. Deze groep omvat myelomonocytaire juveniele leukemie, die representatief is voor beide syndromen (myelodysplastische en myeloproliferatieve). Bijna de helft van de patiënten presenteren met normale of lage neutrofielentellingen en dysplasie van veelvoudige cellijnen zonder organomegalie en beendermerg met morfologie die op vuurvaste bloedarmoede met bovenmatige blasten lijken, maar met monocytose. Andere patiënten hebben ernstige neutrofilia, monocytose en splenomegalie. Het is nog niet bekend of ze 2 verschillende ziekten, een myelodysplastische en een myeloproliferatieve; nochtans, tot nu toe zijn er geen verschillen in cytogenetische abnormaliteiten of in de groeipatronen van de in vitro kolonies of in hun klinische evolutie, zodat is er controverse tussen clinici en pathologen volgens hun plaats binnen de classificatie. Volgens de laatste revisie bevinden zich in deze categorie:
1. Chronische myelomonocytaire leukemie.
2. Atypische chronische myeloïde leukemie (BCR / ABL negatief).
3. Juveniele myelomonocytaire leukemie.
4. Myelodysplastisch / myeloproliferatief syndroom Niet classificeerbaar.
in de categorie acute leukemieën herkent mieloblásticas (LAM) (gedefinieerd als een percentage van meer dan 20% myeloblasten in het beenmerg of bloedverdunner perifeer, of de aanwezigheid van een cytogenetische afwijking in het bijzonder, ondanks het feit dat blasten zijn gemeld) de volgende groepen:
1. LAM met recidiverende cytogenetische translocaties.
2. LAM met myelodysplastische kenmerken.
3. LAM en MDS gerelateerd aan antineoplastische behandelingen.
4. Ik kan niet worden ingedeeld.
5. Myeloïde sarcoom.
6. Myeloïde proliferaties gerelateerd aan het syndroom van Down.
7. Plasmacitoid blastisch neoplasma van dendritische cellen.
lymfoïde neoplasmata
zijn die welke afkomstig zijn van cellen die zich normaliter ontwikkelen tot T-lymfocyten (cytotoxische LT, collaborateurs of regulatoren) of B-lymfocyten (lymfocyten of plasmacellen). In het algemeen worden lymfoïde neoplasmata onderverdeeld in die afgeleid van lymfoïde voorlopers en die van volwassen lymfocyten en plasmacellen en worden vervolgens gegroepeerd volgens hun afstamming (B of T).
historisch gezien zijn lymfoïde neoplasmata die in het beenmerg optreden en waarbij het beenmerg betrokken is, gescheiden van die welke aanwezig zijn als tumor (lymfoom). Nochtans, is het nu bekend dat om het even welk lymfoom met klinische kenmerken van leukemie kan voorstellen en dat om het even welke leukemie af en toe als tumor (granulocytic sarcoom) kan voorstellen. In de WHO-classificatie hangt de diagnose van verschillende lymfoïde neoplasmata niet alleen af van de anatomische locatie van de tumorcellen, maar ook van de morfologisch gedefinieerde oorsprong van de tumorcel. Deze overwegingen hebben de relevantie van de termen L1 en L2 van de FAB-classificatie teniet gedaan, omdat ze niet correleren met hun immunofenotype, genetische afwijkingen of met hun klinisch verloop (Figuur 8). L3 is equivalent aan Burkitt lymfoom in de leukemische fase en moet als zodanig worden gediagnosticeerd.

1. Precursor neoplasmata. Er is een consensus dat precursor neoplasmata die presenteren als solide tumoren en die met beendermerg en bloed biologisch dezelfde ziekte met verschillende klinische presentaties zijn. De meeste precursor lymfoïde neoplasmata worden gepresenteerd als leukemias, dus werd overeengekomen dat de classificatie de term LAL voor de leukemische fase van type b en T precursor neoplasmata moet behouden. Er zijn 2 hoofdcategorieën:
• Precursor B leukemias / lymfomen.
• Precursor leukemias / lymfomen T.
2. Volwassen B-cel neoplasmata. De voorgestelde classificatie beschouwt lymfomen en leukemieën van hetzelfde celtype als dezelfde ziekte met verschillende klinische presentaties of stadia. Specifieke ziekten afkomstig van rijpe B-cellen zijn de volgende:
1. Chronische lymfatische leukemie / klein lymfocyt lymfoom.
2. Lymfoplasmacytisch lymfoom.
3. Mantelcellymfoom.
4. Prolymphocytische B-cel leukemie.
5. Folliculair lymfoom.
6. Diffuus grootcellig lymfoom B.
• intravasculair grootcellig lymfoom B.
• primair mediastinaal grootcellig lymfoom B.
• grootcellig lymfoom B (gerelateerd aan Epstein Barr virus-EBV).
•• Histiocyt-en T-celrijk groot B-cellymfoom.
* diffuus groot B-cellymfoom van het centrale zenuwstelsel.
• * diffuus primair cutaan groot B-cellymfoom.
• diffuus grootcellig B-cellymfoom bij ouderen die positief zijn voor EBV.
• Pasmablastisch lymfoom.
• primair pleuraal lymfoom.
* * alkoompositief (ALK) grootcellig lymfoom.
• Burkitt lymfoom.
7. Marginale zone B-cellymfoom.
8. Extranodaal marginaal zone B-cellymfoom.
9. Milt marginale zone B-cellymfoom
10. Haarcelleukemie.
11. Plasmocytoom / plasmacel myeloom.

myeloïde en lymfoïde LIJNNEOPLASMATA
sommige neoplasmata drukken markers uit van zowel myeloïde als lymfoïde lijnen.ongedifferentieerde) of huidige kenmerken van beide lijnen (gemengd fenotype of gemengde lijn acute leukemie).
Tabel 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍA
Bassan R, Hoelzer D. moderne therapie van Acute lymfoblastische leukemie. J Clin Oncol. 2011;29:523-43.
Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in Acute leukemie. J Clin Oncol. 2011;29:487-94.
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasmas and beyond: evoluing concepts and practical applications. Bloed. 2011 May 12; 117(19): 5019-32. Epub 2011 7 Feb. Beoordeling.
Cortes J, Hochhaus A, Hugues T, Kantajian H. Frontline-en Salvagetherapieën met tyrosinekinaseremmers en andere behandelingen bij chronische myeloïde leukemie. J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologie, risico stratificatie, en therapie van pediatrische scherpe leukemie: een Update. J Clin Oncol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado Mr. Multicenter independent assessment of ourcomes in chronic myeloid leukemia patients treated with Imatinib. Tijdschrift National Cancer Institute. 2011;103:1-9.
Grever M, Lozanki G. Moderne strategieën voor haarcelleukemie. J Clin Oncol. 2011:29;583-90.
Gribben JG, O ‘ Brien S. Update on Therapy of Chronic Lymphocytic Leukemia. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Chronische Myeloïde Leukemie. Huidige concepten in de fysiopathologie en behandeling. Cancerología. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib vergeleken met Imatinib/Cytarabine voor de eerstelijnsbehandeling van vroege Philadelphia chromosoom positieve chronische myeloïde leukemie. Resultaten van een gerandomiseerde klinische studie van de Mexicaanse collaboratieve leukemie Groep. Klinische Leukemie. 2008: 2(2);1128-32.
Lichtman MA. Classificatie en klinische manifestaties van de klonale myeloïde aandoeningen. Nl: Williams. Hematologie. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Molecular Genetics of adult acute myeloid leukemie. Prognostische en therapeutische implicaties. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rev Fac Med UNAM. 2010; 53:36-43.
Sanz M, Lo-Coco F. moderne benaderingen voor de behandeling van Acute Promyelocytaire leukemie. J Clin Oncol. 2011;29:495-503.
opmerking
* dysplasie. Het verwijst naar de cytomorfologische verandering die dissociatie van nucleus-cytoplasma rijping omvat (vergeet niet dat chromatin rijping afhangt van DNA en RNA cytoplasma, daarom stopt de kern bij rijping terwijl het cytoplasma zijn normale proces voortzet) die niet-levensvatbare cellen produceert en er intramedullaire apoptose is.