gjennomgangsartikkel
Leukemi-for fastlegen
Leukemi for fastlegen
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
Leder av institutt For Hematologi. Sykehus Á del Pedregal. Mexico, DF. E-postadresse: [email protected]
B Intern Medisin. Sykehus Á del Pedregal. Mexico. DF.
C Intern Medisin. Sykehus Á del Pedregal. Mexico. DF.
Mottatt: 17. oktober 2011
Akseptert: 07 januar 2012
INNLEDNING
Til tross for de store molekylære og terapeutiske fremskrittene i studiet av leukemier, er de grunnleggende aspektene ved denne tilstanden ennå ikke klart kjent av ikke-hematologen, så målet med dette arbeidet er å gi grunnleggende informasjon til medisinske studenter og leger generelt, og det tillater fremfor alt å få generell kunnskap om leukemier, deres rettidige diagnose og søke tidlig referanse med hematologen.

Definisjon
Leukemi Er begrepet som brukes til å definere en gruppe ondartede blodsykdommer. Tidlig diagnose er viktig, da det vil tillate pasienten å gå tidlig med hematologispesialisten, som vil lede diagnoseprosessen og tilby den spesifikke behandlingen. Det er preget av å ha en klonal, autonom og unormal proliferasjon av cellene som gir opphav til resten av blodets normale celler (tumoradferd generelt). Dette innebærer at en tidlig celle gjennomgår en genetisk forandring som vil føre til at en unormal klon (koloni) av seg selv oppstår ukontrollert. Denne unormale produksjonen er uordnet fordi de unormale cellene multipliserer i bilde og likhet av seg selv, slik at de gradvis opptar plass i det normale benmarget og forårsaker progressiv anemi, unormal blødning og predisponering for infeksjoner. På den annen side, når unormale celler invaderer andre vev, vil det være svikt i det aktuelle organets funksjon, for eksempel infiltrering til sentralnervesystemet som oppstår ved akutt lymfoblastisk leukemi (LAL) kan manifestere seg med hodepine, anfall, fokuserte motorendringer, økt intrakranielt trykk og manglende tidlig diagnose og tilstrekkelig behandling, vil presentere tap av funksjon og irreversible konsekvenser.
KLINISKE MANIFESTASJONER
det kliniske bildet er variert og avhenger av typen leukemi: akutt eller kronisk, men for 2 er det ikke-spesifikke kliniske manifestasjoner (som forekommer i noen sykdom):
1. Tretthet.
2. Lett tretthet.
3. Generell svakhet.
4. Ønsker å forbli hvile eller i sengen.
5. Det krever hjelp fra noen til å møte dine personlige behov.
Kroniske leukemier er indolente og opptil 50% av tilfellene oppdages i en rutinemessig klinisk eller laboratorievurdering hos frivillige som anses å være sunne og kommer til å donere blod, men når sykdommen utvikler seg, presenteres ikke-spesifikke manifestasjoner, men nå er de spesifikke (Tabell 1).

i akutte former er spesifikke manifestasjoner avledet fra mangel på en av cellelinjene:
1. Erytrocytter: anemisk syndrom hvis intensitet vil avhenge av graden av hypoksemi uavhengig av graden av anemi. Dyspnø av middels anstrengelse til orthoprea.
2. Blodplater: petechiae, ekkymose i ekstremiteter, og i mer alvorlige generaliserte tilfeller, tørr og våt blødning med epistaxis, gingivorrhagia, hematuri, mane eller hematokesi. Svært alvorlig i sentralnervesystemet (CNS).
3. Leukocytter: feber, diaphorese, lokaliserte infeksjoner opp til frank septikemi(bakterier eller sopp). De forekommer med nøytropeni mindre enn 250 totale nøytrofiler / mm3.
Infiltrativ syndrom: refererer til unormal implantasjon i noe vev, selv om det er vanlig:
1. Hepatomegali eller splenomegali (Figur 5).
2. Adenomegali(lokal eller generalisert).
3. Leukemisk hudfarge.
4. Bone smerte fra beinmarg ekspansjon.
5. Myke vev(granulocytisk sarkom).
6. Testikkel.
7. SNC.
8. Tannkjøtt og ethvert sted (figur 1).

Metabolske forstyrrelser: de skyldes unormal hyperproduksjon av ondartede celler og økt apoptose.
1. Acidose.
2. Økt melkesyre dehydrogenase (LLD).
3. Hyperkalemi.
4. Hyperurikemi.
5. Økt β 2 mikroglobulin.

det kliniske beviset dominerer som hjørnesteinen i de mistenkte diagnosene av akutte leukemier og av enhver tilstand, men det som følger er å utfylle diagnosen med støtte fra det kliniske laboratoriet i cytometri, komplett blodtelling, eller spesielt, det vil si den grundige observasjonen av perifert blodutstryk av det tekniske personalet som har diagnosen.forberedelse i identifisering av unormale celler, spesielt leukemi.

Laboratorieendringer som krever spesiell gjennomgang inkluderer:
1. Anemi (hvilken som helst klasse).
2. Leukopeni eller leukocytose(overvekt av en cellelinje).
3. Trombocytopeni.
4. Kombinasjoner: bicitopeni eller pankytopeni.
spesiell forsiktighet bør utvises når laboratoriet rapporterer tilstedeværelse av leukocytter eller atypiske lymfocytter(kan være leukemiske blaster). Det anbefales å be om en ekspertanmeldelse(Figur 2).

benmargsaspirasjon er viktig for diagnose (Figur 3) og 20% av blastene kreves for å etablere kriteriene for akutt leukemi i noen av dens varianter.

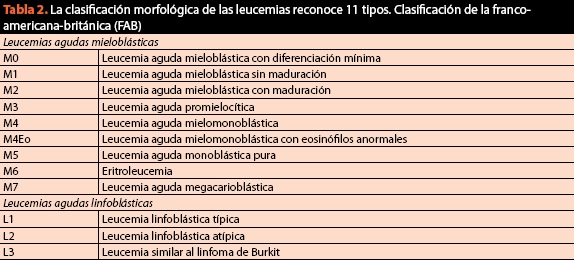
i samme prosedyre skal det hentes prøver for den endelige klassifiseringen av tilstanden og be om karyotype og immunfenotype, siden det cytomorfologiske kriteriet for tiden er av avgjørende betydning, men ikke lenger er tilstrekkelig. Nåværende og nåværende klassifisering av ondartede blodsykdommer er oppført i Tabell 2.
behandlingen er rettet mot 2 viktige aspekter: den første av dem er den spesifikke antileukemiske og er basert på bruk av legemidler av kjemisk opprinnelse som er kjent som kjemoterapi, hvis hovedmål er å utrydde, det vil si eliminere alle leukemiske celler fra kroppen. Det andre aspektet av behandlingen er støtte for komplikasjoner som vanligvis oppstår hos pasienter ved opptak.
behandlingen er rettet mot 2 viktige aspekter: den første er den spesifikke antileukemiske og er basert på bruk av legemidler av kjemisk opprinnelse som er kjent som kjemoterapi, hvis hovedmål er å utrydde, det vil si eliminere alle leukemiske celler fra kroppen.
det andre aspektet av behandlingen er støtten til komplikasjonene som pasientene vanligvis presenterer ved opptak, for eksempel:
1. Anemi.
2. Unormal blødning.
3. Lunge-og generaliserte infeksjoner, Blant Annet (Figur 4).
4. Eventuelle andre tilstøtende komplikasjoner som pasienten kan ha( komorbiditet), for eksempel eksisterende forhold, for eksempel diabetes, hypertensjon, hjertesykdom og andre sykdommer som er vanlige blant pasienter som lider av leukemi.


derfor er det svært viktig å ta hensyn til at behandlingen mot leukemi er tverrfaglig, med deltagelse av andre spesialister som støtte til hematologen.
Antileukemisk behandling vil også være forskjellig for ulike typer leukemi og for akutte former. Den er delt inn i 3 faser:
1. Induksjon av remisjon. Målet er å oppnå fullstendig remisjon( CR), det vil si normalisering av pasientens blodverdier, fraværet av symptomer eller tegn på at leukemi vedvarer med infiltrering. Under prosessen skal pasienten ha en «leukemifri tilstand» i benmargen, og fremtiden skal være utvinningen til en normal hematopoiesis, og dessverre i andre tilfeller gjenoppretter de med sykdommen, som taler om resistent leukemi hvis prognose er forferdelig. Denne første prosessen kan ta 6 til 8 uker for å oppnå CR.
2. Konsolidering. Det innebærer bruk av de samme stoffene som ble brukt i induksjon eller kombinasjon av andre kjemoterapeutika, også med det formål å følge utryddelsen av resterende maligne celler som kan utvikle motstand mot førstegangsbruk.
3. Vedlikehold. Det er foretrukket å holde pasienten under effekten av kjemoterapi før muligheten for begynnende leukemisk aktivitet, og at den ved behandlingen opprettholder effekten til sykdommen forsvinner.

Så langt for akutte former blir de strengeste kriteriene for å vurdere CR blitt mer komplekse, siden det er best å lete etter molekylær remisjon, som innebærer søket etter den første kromosomale endringen som forekommer ved promyelocytisk leukemi (m3-FAB) med t (15;17) initial, det bør søkes etter spesifikke molekylære og cytogenetiske studier til tross for å være I CR, siden hvis translokasjonen vedvarer, bør den aggressive behandlingen fortsette til total eliminering av klonen. Denne variasjonen av leukemi bør betraktes som potensielt herdbar og er en Av De vanligste I Latino-befolkningen.
helbredelsen av sykdommen vil da avhenge av eliminering av alle eksisterende maligne celler i pasienten. Generelt kan enkelte leukemier være utsatt for kur med kjemoterapi alene, men i dag må det legges stor vekt på såkalte prognostiske faktorer som er basert på matematiske modeller som gjør det mulig for pasienter å bli plassert i prognosegraden de har og inkluderer:
1. Typen av leukemi.
2. Den første molekylære endringen og dens utholdenhet til tross for behandling eller utryddelse.
3. alder. Pasienter eldre enn 60 år har en dårlig prognose sammenlignet med yngre pasienter.
4. Kjemoterapi. Legemidlene angitt og fremfor alt de anbefalte dosene skal brukes. For eksempel, i voksen LAL HyperCyVAD skjema (eskalerte doser av cyklofosfamid, vinkristin, adriamycin, deksametason i kombinasjon med cytosin arabinosid og metotreksat) oppnår 90% CR og kurer i 50% av tilfellene, data som ikke tidligere er sett med andre ordninger. Denne behandlingen er giftig og krever at den bare brukes i institusjoner som har tilstrekkelige støtteressurser. Dessverre, i vårt miljø har ikke alle sentre de anbefalte medisinene, og resultatene vil ikke være reproduserbare, siden de ikke har nok støtte til pasienten i fasen med maksimal myelosuppresjon.
5. Støttende terapi. Resultatene av kjemoterapi og nye stoffer, nye kombinasjoner og med mer spesifisitet tvinger implementeringen av de tverrfaglige lagene med adressen til hematologen; installasjon og bruk av sentrale venekatetre; støtte til blodbank for støtte av transfusjoner av blodplater og erytrocytter (selv produkter stellat); inngrep av infektologen for påvisning av infeksjoner og riktig bruk av antibiotika eller antifungale midler, enten profylaktisk eller terapeutisk, om nødvendig; tilveiebringelse av isolasjonsrom med vedlikehold og quartermaster-tjenester som gjør det mulig å oppnå et bakteriefritt miljø, inkludert steril tilførsel; et laboratorium med pasientprøvehåndteringsprotokoll i spesifikke behandlingsprotokoller og styring av spesielle prøver (forberedelse av leukocyttkonsentrater i buffy coat hematisk cytometri og oppnå optimal avlesning); på den annen side, er det viktigste sykepleie, hjelpe-og administrativt personell, som med hele gruppen er forventet resultatene de har i andre land.
6. Benmargstransplantasjon (MO). Det er en kompleks og høy pris type behandling som krever en kompatibel MO donor og en inaktiv tilstand med høy sannsynlighet for tidlig eller sen tilbakefall eller, med faktorer med dårlig prognose. Det er en prosedyre med mer kurative tendenser siden den bruker megadoser av kjemoterapi for å utrydde leukemiske celler, men i forsøket utrydder den også de normale forløperne, og erstatning av en ny kompatibel normal marg er nødvendig. Typen allogen (søskenkompatibel identisk) er det beste valget siden mottakeren aksepterer bare ved identitet delvis( den eneste av total aksept er identiske tvillinger), og resulterer derfor i en avvisning av graft-versus-host-sykdommen, som igjen fører til graft-versus-leukemi og øker utryddelsen av klonene leukemi, men i alvorlige tilfeller av syndromet (grad 3-4) øker morbiditeten og dødeligheten til tross for den spesifikke håndteringen.
KRONISK LEUKEMI
1. Kronisk lymfocytisk leukemi. Det forekommer oftere hos eldre mennesker, og kriteriet er persistensen av lymfocytose på mer enn 10 x 109 / l, OG MO med infiltrering av mer enn 50% lymfocytter MED CD5 + fenotype. Kriteriet for behandling er duplisering av kontoen av lymfocytter i et år eller progresjon av adenomegalia eller splenomegali, selv om noen tilfeller utover denne standarden ved tilstedeværelse av hemolytisk anemi eller trombocytopeni, autoimmun, og deretter indikert behandling basert på kombinasjonen av fludarabin, cyklofosfamid, og prednison, i trinn i OG II bare somenter og observert og forventet utvikling uten behandling.
2. Kronisk myelogen leukemi (KML). I denne sykdommen er det et stort fremskritt i kunnskapen om Tilstedeværelsen Av Philadelphia-kromosomet, som ble beskrevet i 1950 i Byen American Union, og at begynnelsen var det første markørkromosomet i forbindelse med malignitet, men i løpet av årene med forskning klarte vi å kjenne t (9;22) med funksjonell ekspresjon av kromosomet med produksjon av et onkoprotein med stor tyrokinokinaseaktivitet, noe som øker celleproliferasjon og som igjen forklarer den store leukocytose og trombocytose som disse pasientene presenterer, samt den store splenomegali (Figur 5).
På dette tidspunktet har 10 til 12 år gått siden oppdagelsen av et lite molekyl spesielt rettet mot dette fosfat-donormolekylære substratet for intern regulering av leukemisk celle og dens substrater. DEN opprinnelig kalt STI (signaltransduksjonshemmer) produserte konkurransedyktig inhibering av fosforyleringen, førte til celleapoptose og pasientene oppnådde kliniske resultater som aldri før ble sett med molekylære remisjoner på opptil 80-90% ved 10 år, noe som dramatisk endret sykdommens naturlige historie, med forbehandlingen ikke var mer enn 3 år. Denne informasjonen gjør en endring i naturhistorie og prognose av sykdommen, tidligere dødelig på kort sikt.

disse eksemplene er en base av fremdriften så intens som skjedde de siste årene, og At det er ønskelig å tillate provosere interessen til medisinske studenter, deres lærere, men spesielt av utdanningsmyndighetene i leukemi generelt, som opptar de første 5 stedene av frekvensen av ondartet sykdom hos voksne og de første stedene i barna, slik at hematologien skal inngå i kjernefagene i læreplanen for medisinens karriere og til og med som en del av kursene videreutdanning spesialisering. Selv om det er nyttig for sin enkelhet, Kan Den Britiske Fransk-Amerikanske klassifiseringen (FAB) føre til diagnostiske og derfor terapeutiske feil i opptil 20% av tilfellene. Av denne grunn har klassifisering ved immunhistokjemi og molekylærbiologiske metoder blitt et ikke-krav for riktig klassifisering og påfølgende behandling av pasienter (Figur 6).

i motsetning til klassifiseringen FAB, REFLEKTERER WHO (tabell 2) en endring i paradigmet som vi forstår til sykdommer blod, fordi for første gang kombinert genetisk informasjon, morfologiske, cytokjemiske og immunfenotypiske med de kliniske funnene innenfor de diagnostiske algoritmer for neoplasmer i hematopoietisk vev; den relative betydningen av hvert kriterium varierer mellom neoplasmer og det er ingen «gullstandard» for klassifisering av alle hematologiske maligniteter. Målet var å definere enheter som kunne gjenkjennes av patologer og som hadde klinisk relevans. Siden oppstarten i 2001 har det blitt gjort ulike revisjoner for å oppdatere innholdet i forhold til de nyeste funnene. Den siste gjennomgangen var fra 2008 og klassifiserer hematologiske maligniteter som følger:
1. Myeloide neoplasmer.
2. Lymfoide neoplasmer.
3. Mastcelle eller fettcellesykdommer.
4. Histiocytiske og dendritiske cellesykdommer.

NEOPLASIER MYELOIDE
MYELOIDE neoplasmer er avledet fra stamceller i benmargen, differensieres til erytrocytter, granulocytter (nøytrofiler, basofiler og eosinofiler), monocytter og megakaryocytter. FAB-klassifiseringen gjenkjenner 3 hovedkategorier:
1. Akutt myelogen leukemi.
2. Myelodysplastiske syndromer.
3. Myeloproliferative neoplasmer.
de viktigste determinanter av kategoriene anerkjent ved hjelp av morfologiske, histokjemiske og immunfenotypiske og prosentandelen blastceller, cellelinjen og graden av differensiering av neoplastiske celler (figur 7). De siste årene har de genetiske egenskapene (cytogenetiske og molekylære), samt forbehandlingen og utviklingen av myelodysplasi, vist en betydelig innvirkning på den kliniske oppførselen til disse lidelsene, som ikke alltid korrelerer godt med KATEGORIENE AV FAB, slik at den sentrale debatten for omklassifisering var å diskriminere mellom enhetene og patologiske prognostiske faktorer, for å oppnå en klassifisering med klinisk relevans og betydning for patologen. Noen genetiske avvik synes å definere ulike sykdommer, mens andre representerer prognostiske faktorer for en bestemt sykdom.

FOR tiden GRUPPERER WHO-klassifikasjonen myeloide tilstander i 4 hovedgrupper:
1. Myeloproliferative sykdommer
2. Myelodysplastisk syndrom
3. Myelodysplastiske / myeloproliferative sykdommer
4. Akutt myeloid leukemi

Myeloproliferative sykdommer er en gruppe klonale lidelser assosiert med spredning av en eller flere myeloide linjer. Det blir stadig tydeligere at disse sykdommene ofte er forbundet med mutasjoner som forårsaker unormale økninger i tyrosinkinaseaktivitet og vekstfaktoruavhengig progenitorcelleproliferasjon i benmargen. Prosentandelen av blaster i beinmarg er normal eller litt høy, men alltid mindre enn 20%. Hematopoiesis er vanligvis effektiv, noe som resulterer i en økning i tellingen av en eller flere modne celler i perifert blod. Prototypen av myeloproliferative neoplasmer er Philadelphia kromosom positiv (Ph1) kronisk myeloid leukemi (BCR/ABL). De andre enhetene som er inkludert er:
1. Polycytemi vera.
2. Idiopatisk myelofibrose.
3. Primær essensiell trombocytemi.
4. Kronisk eosinofil leukemi.
5. Kronisk nøytrofil leukemi.
6. Mastocytose.
7. Uklassifiserbare myeloproliferative neoplasmer.

Myelodysplastiske syndromer refererer til lidelser preget av ineffektiv celleproduksjon og dysplasi, med en variabel risiko for transformasjon til akutt leukemi. Cellularitet i margen er ofte økt, men svært variabel. Det er modning, men også dysplasi av en eller flere myeloide linjer. Hematopoiesis er ikke effektiv og derfor eksisterer cytopenier. Dette elementet inkluderer:
1. Refraktær cytopeni med en-linje dysplasi*.
• Refraktær anemi.
• Refraktær nøytropeni.
• Refraktær trombocytopeni.
2. Ildfast anemi med ringformede sideroblaster.
3. Refraktær cytopeni med dysplasi av flere linjer.
4. Ildfast anemi med overskytende blaster.
5. Myelodysplastisk syndrom med d (5q).
6. Uklassifiserbart myelodyspastisk syndrom.
7. Juvenil myelodysplastisk syndrom, inkluderer en foreløpig enhet kjent som juvenil refraktær cytopeni.
Myelodysplastiske / myeloproliferative syndromer inkluderer lidelser der dysplastiske og proliferative egenskaper sameksisterer. Denne gruppen inkluderer myelomonocytisk juvenil leukemi, som er representativ for begge syndromer(myelodysplastisk og myeloproliferativ). Nesten halvparten av pasientene er tilstede med normale eller lave nøytrofiltall og dysplasi av flere cellelinjer uten organomegali og benmarg med morfologi som ligner ildfast anemi med overskytende blaster, men med monocytose. Andre pasienter har alvorlig nøytrofili, monocytose og splenomegali. Det er ennå ikke kjent om de er 2 forskjellige sykdommer, en myelodysplastisk og en myeloproliferativ; men så langt er det ingen forskjeller i cytogenetiske abnormiteter eller i vekstmønstrene til in vitro-koloniene eller i deres kliniske utvikling, så det er kontrovers mellom klinikere og patologer i henhold til deres plass i klassifiseringen. Ifølge den siste revisjonen ligger i denne kategorien:
1. Kronisk myelomonocytisk leukemi.
2. Atypisk kronisk myelogen leukemi (bcr / ABL negativ).
3. Juvenil myelomonocytisk leukemi.
4. Myelodysplastisk / myeloproliferativ syndrom kan ikke klassifiseres.
i kategorien akutt leukemi mieloblá (LAM) (som er definert med en prosentandel større enn 20% myeloblaster i benmargen eller blod tynnere perifer, eller tilstedeværelsen av en cytogenetisk abnormitet spesielt, til tross for hensyn til blaster) gjenkjenner følgende grupper:
1. LAM med tilbakevendende cytogenetiske translokasjoner.
2. LAM med myelodysplastiske egenskaper.
3. LAM og MDS relatert til antineoplastiske behandlinger.
4. LAM ikke klassifiserbar.
5. Myeloid sarkom.
6. Myeloide proliferasjoner relatert Til Downs syndrom.
7. Plasmacitoid blastisk neoplasma av dendritiske celler.
LYMFOIDE NEOPLASMER
ER de som stammer fra celler som normalt utvikler Seg Til T-lymfocytter (cytotoksisk LT, samarbeidspartnere eller regulatorer) eller B-lymfocytter (lymfocytter eller plasmaceller). Generelt er lymfoide neoplasmer delt inn i de som er avledet fra lymfoide forløpere og de fra modne lymfocytter og plasmaceller og grupperes deretter i henhold til deres avstamning (B eller T).
historisk sett har lymfoide neoplasmer som forekommer i beinmarg og involverer beinmarg blitt skilt fra de som presenterer som en svulst (lymfom). Imidlertid er det nå kjent at ethvert lymfom kan presentere med kliniske trekk ved leukemi, og at enhver leukemi noen ganger kan presentere som en svulst (granulocytisk sarkom). I WHO-klassifiseringen avhenger diagnosen av flere lymfoide neoplasmer ikke bare av tumorcellens anatomiske plassering, men også på tumorcellens morfologisk definerte opprinnelse. Disse hensynene opphevet relevansen Av begrepene L1 Og L2 I FAB-klassifiseringen, siden de ikke korrelerer med deres immunfenotype, genetiske abnormiteter eller med deres kliniske forløp (Figur 8). L3 er ekvivalent Med Burkitt lymfom i leukemisk fase og bør diagnostiseres som sådan.

1. Forløper neoplasmer. Det er enighet om at forløpernes neoplasmer som presenterer som faste svulster og de som involverer benmarg og blod, er biologisk den samme sykdommen med forskjellige kliniske presentasjoner. De fleste forløperlymfoide neoplasmer er presentert som leukemier, så det ble avtalt at klassifiseringen skulle beholde termen LAL for leukemisk fase av type B og T forløpernoplasmer. Det er 2 hovedkategorier:
• Forløper b leukemier / lymfomer.
• forløper leukemi/lymfom T.
2. Modne b-celle neoplasmer. Den foreslåtte klassifiseringen vurderer lymfomer og leukemier av samme celletype som samme sykdom med ulike kliniske presentasjoner eller stadier. Spesifikke sykdommer avledet fra modne b-celler er som følger:
1. Kronisk lymfocytisk leukemi / lite lymfocyttlymfom.
2. Lymfoplasmacytisk lymfom.
3. Mantelcellelymfom.
4. Prolymphocytisk b-celle leukemi.
5. Follikulært lymfom.
6. Diffus storcellelymfom B.
• Intravaskulær storcellelymfom B.
• Primær mediastinal storcellelymfom B.
* storcellelymfom B (relatert Til Epstein Barr virus-EBV) •
•• Histiocytt-og t-cellerikt stort b-cellelymfom.
* Diffust sentralnervesystemet stort b-celle lymfom.
• * Diffus primært kutant stort b-celle lymfom.
• Diffus stort b-cellelymfom hos eldre positivt FOR EBV.
• Pasmablastisk lymfom.
• Primær pleural lymfom.
• * Alkoma-positivt (ALK) stort celle lymfom. Burkitt lymfom.
7. Marginal sone B-celle lymfom.
8. Ekstranodal marginal sone B-celle lymfom.
9. Milt marginal sone B-celle lymfom
10. Hårete celle leukemi.
11. Plasmocytom / plasmacelle myelom.

MYELOIDE OG LYMFOIDE LINJER uttrykker markører for både myeloide og lymfoide linjer.begge linjer (blandet fenotype eller blandet linje akutt leukemi).
Tabell 3.Classificaci hryvnias de la oms de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍ
Bassan R, Hoelzer D. Moderne Terapi Av Akutt Lymfoblastisk Leukemi. J Clin Oncol. 2011;29:523-43.
Burnett A, Wetzler M, Lö B. Terapeutiske fremskritt I Akutt Leukemi. J Clin Oncol. 2011;29:487-94.
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. 2008 WHO klassifisering av lymfoide neoplasmer og utover: utviklende konsepter og praktiske anvendelser. Blod. 2011 Mai 12;117 (19): 5019-32. Epub 2011 Februar 7. Anmeldelse. Cortes J, Hochhaus A, Hugues T, Kantajian H. Front Line Og Salvage Terapi Med Tyrosinkinasehemmere og andre Behandlinger I Kronisk Myelogen Leukemi. J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologi, Risikostratifisering Og Terapi Av Pediatrisk Akutt Leukemi: En Oppdatering. J Clin Oncol. 2011;29:551-65. Gambacorti PC, Antolin L, Hurtado Mr. Multicenter uavhengig vurdering av ourcomes hos pasienter med kronisk myelogen leukemi behandlet Med Imatinib. Tidsskriftet National Cancer Institute. 2011;103:1-9.
Grever M, Lozanki G. Moderne Strategier For Hårete Celle Leukemi. J Clin Oncol. 2011:29;583-90.
Gribben JG, O ‘ Brien S. Oppdatering Om Behandling Av Kronisk Lymfocytisk Leukemi. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Kronisk Myelogen Leukemi. Aktuelle Begreper I Fysiopatologi og Behandling. Cancerologí. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib sammenlignet Med Imatinib/Cytarabin for førstelinjebehandling av Tidlig Philadelphiakromosom positiv Kronisk Myelogen leukemi. Resultater Av En Randomisert klinisk studie Av Den Meksikanske Collaborative Leukemia Group. Klinisk Leukemi. 2008: 2(2);1128-32.
Lichtman MA. Klassifisering og kliniske manifestasjoner av klonale myeloide lidelser. En: Williams. Hematologi. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Molekylær Genetikk av voksen akutt myeloid Leukemi. Prognostiske Og Terapeutiske Implikasjoner. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiolog@a De La Citometrí Hemá Rev Fac MED UNAM. 2010; 53:36-43. Sanz M, Lo-Coco F. Moderne Tilnærminger til behandling Av Akutt Promyelocytisk Leukemi. J Clin Oncol. 2011;29:495-503.
Merk
* Dysplasi. Det refererer til den cytomorfologiske endringen som inkluderer dissosiasjon av kjerne-cytoplasma modning (husk at kromatin modning avhenger AV DNA og rna cytoplasma, derfor stopper kjernen ved modning mens cytoplasma fortsetter sin normale prosess) som produserer ikke-levedyktige celler og det er intramedullær apoptose.