recensione Articolo
Leucemia-per il medico di medicina generale
Leucemia per il medico di medicina generale
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
Capo del dipartimento di Ematologia. OspedalegGeles del Pedregal. Messico, DF. Indirizzo email: [email protected]
b Medicina interna. OspedalegGeles del Pedregal. Messico. DF.
c Medicina interna. OspedalegGeles del Pedregal. Messico. DF.
Ricevuto: 17 ottobre 2011
Accettato: 07 gennaio 2012
INTRODUZIONE
Nonostante il grande molecolare e progressi terapeutici nello studio delle leucemie, gli aspetti fondamentali di questa condizione non sono ancora ben conosciuti dai non-ematologo, quindi, l’obiettivo di questo lavoro è quello di fornire informazioni fondamentali per gli studenti di medicina e medici in generale, e che permette di sopra di tutti per ottenere una conoscenza generale di leucemie, la loro tempestiva diagnosi e cercano di riferimento iniziale con l’ematologo.

DEFINIZIONE
Leucemia è il termine usato per definire un gruppo di malattie del sangue maligne. La diagnosi precoce è essenziale, in quanto consentirà al paziente di andare presto con lo specialista di ematologia, che guiderà il processo diagnostico e offrirà il trattamento specifico. È caratterizzato dall’avere una proliferazione clonale, autonoma e anormale delle cellule che danno origine al resto delle cellule normali del sangue (comportamento tumorale in generale).
Ciò implica che una cellula precoce subisce un cambiamento genetico che causerà un clone anormale (colonia) di se stesso a verificarsi in modo incontrollabile. Questa produzione anormale è disordinata perché le cellule anormali si moltiplicano a immagine e somiglianza di se stesse, quindi occupano gradualmente lo spazio del midollo osseo normale e causano anemia progressiva, sanguinamento anormale e predisposizione alle infezioni. D’altra parte, quando le cellule anormali invadere altri tessuti, ci sarà il fallimento di funzionamento dell’organo in questione, per esempio, infiltrazione del sistema nervoso centrale che si verifica nella leucemia linfoblastica acuta (LAL) potrebbe manifestarsi con mal di testa, convulsioni, focalizzata motore alterazioni, aumento della pressione intracranica, e la mancata effettuazione di una diagnosi precoce e fornire un trattamento adeguato, presenterà la perdita di funzione e di conseguenze irreversibili.
MANIFESTAZIONI CLINICHE
Il quadro clinico è vario e dipende dal tipo di leucemia: acuta o cronica, tuttavia per l ‘ 2 ci sono manifestazioni cliniche non specifiche (che si verificano in qualsiasi malattia):
1. Fatica.
2. Facile stanchezza.
3. Debolezza generalizzata.
4. Desidera rimanere a riposo o a letto.
5. Richiede l’aiuto di qualcuno per soddisfare le vostre esigenze personali.
Le leucemie croniche sono indolenti e fino al 50% dei casi vengono scoperti in una revisione clinica o di laboratorio di routine in volontari che sono considerati sani e vengono a donare il sangue, tuttavia, con il progredire della malattia, vengono presentate manifestazioni non specifiche ma ora sono specifiche (Tabella 1).

Nelle forme acute, le manifestazioni specifiche derivano dalla carenza di una delle linee cellulari:
1. Eritrociti: sindrome anemica la cui intensità dipenderà dal grado di ipossiemia indipendentemente dal grado di anemia. Dispnea di sforzo medio fino all’ortoprea.
2. Piastrine: petecchie, ecchimosi alle estremità, e nei casi generalizzati più gravi, emorragia secca e umida con epistassi, gengivorragia, ematuria, criniera o ematochesia. Molto grave nel sistema nervoso centrale (SNC).
3. Leucociti: febbre, diaforesi, infezioni localizzate fino a setticemia franca (batteri o funghi). Si verificano con neutropenia inferiore a 250 neutrofili totali / mm3.
Sindrome infiltrativa: si riferisce all’impianto anormale in qualsiasi tessuto, sebbene sia comune:
1. Epatomegalia o splenomegalia (Figura 5).
2. Adenomegalia (locale o generalizzata).
3. Carnagione leucemica.
4. Dolore osseo da espansione del midollo osseo.
5. Tessuti molli (sarcoma granulocitico).
6. Testicolo.
7. SNC.
8. Gengive e qualsiasi sito (figura 1).

Disordini metabolici: derivano da un’iperproduzione anormale di cellule maligne e da un aumento dell’apoptosi.
1. Acidosi.
2. Aumento della lattica deidrogenasi (LLD).
3. Iperkaliemia.
4. Iperuricemia.
5. Aumento della microglobulina β2.

L’evidenza clinica predomina la pietra angolare della sospetta diagnosi di leucemie acute e di qualsiasi condizione, ma ciò che segue è quello di integrare la diagnosi con il supporto del laboratorio clinico in citometria a conteggio completo del sangue, o speciale, che è a dire, l’accurata osservazione dello striscio di sangue periferico da personale tecnico che ha preparato l’identificazione di cellule anomale, in particolare la leucemia.

Le alterazioni di laboratorio che richiedono una revisione speciale includono:
1. Anemia (qualsiasi grado).
2. Leucopenia o leucocitosi (predominanza di una linea cellulare).
3. Trombocitopenia.
4. Combinazioni: bicitopenia o pancitopenia.
Si deve prestare particolare attenzione quando il laboratorio segnala la presenza di leucociti o linfociti atipici (possono essere blasti leucemici). Si consiglia di richiedere una perizia (Figura 2).

L’aspirazione del midollo osseo è essenziale per la diagnosi (Figura 3) e il 20% delle esplosioni è necessario per stabilire i criteri per la leucemia acuta in una qualsiasi delle sue varietà.

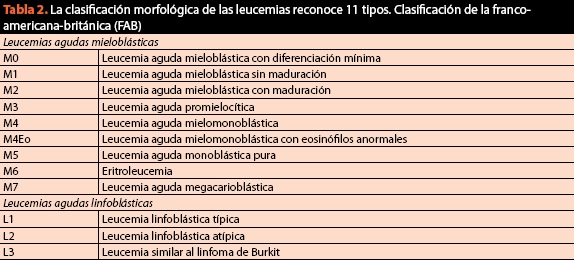
Nella stessa procedura si devono ottenere campioni per la classificazione finale della condizione e richiedere cariotipo e immunofenotipo, poiché attualmente il criterio citomorfologico è di vitale importanza ma non è più sufficiente. La classificazione attuale e attuale delle malattie del sangue maligne è elencata nella Tabella 2.
Il trattamento si rivolge a 2 aspetti importanti: il primo di essi è l’antileucemico specifico e si basa sull’uso di farmaci di origine chimica noti come chemioterapia, il cui obiettivo principale è quello di sradicare, cioè eliminare tutte le cellule leucemiche dal corpo. Il secondo aspetto del trattamento è il supporto per le complicanze che di solito si verificano nei pazienti al momento del ricovero.
Il trattamento si rivolge a 2 aspetti importanti: il primo di essi è l’antileucemico specifico e si basa sull’uso di farmaci di origine chimica noti come chemioterapia, il cui obiettivo principale è quello di sradicare, cioè eliminare tutte le cellule leucemiche dal corpo.
Il secondo aspetto del trattamento è il supporto per le complicanze che i pazienti di solito presentano al momento del ricovero, come ad esempio:
1. Anemia.
2. Sanguinamento anormale.
3. Infezioni polmonari e generalizzate, tra le altre (Figura 4).
4. Eventuali altre complicazioni adiacenti che il paziente può avere (comorbilità), come condizioni preesistenti, ad esempio diabete, ipertensione, malattie cardiache e altre malattie comuni tra i pazienti affetti da leucemia.


pertanto, è molto importante prendere in considerazione che il trattamento contro la leucemia è multidisciplinare, che prevedono la partecipazione di altri specialisti, come il supporto per l’ematologo.
Il trattamento antileucemico sarà anche diverso per diversi tipi di leucemia e per forme acute. È diviso in 3 fasi:
1. Induzione della remissione. L’obiettivo è raggiungere la remissione completa (CR), cioè la normalizzazione dei valori ematici del paziente, l’assenza di sintomi o segni che la leucemia persiste con infiltrazione. Durante il processo il paziente dovrebbe avere uno “stato libero da leucemia” nel midollo osseo e il futuro dovrebbe essere il recupero di una normale emopoiesi, e purtroppo in altri casi, si riprendono con la malattia, che parla di leucemia resistente la cui prognosi è terribile. Questo primo processo può richiedere da 6 a 8 settimane per ottenere CR.
2. Consolidamento. Prevede l’uso degli stessi farmaci che sono stati utilizzati nell’induzione o combinazione di altri chemioterapici, anche allo scopo di seguire l’eradicazione di cellule maligne residue che potrebbero sviluppare resistenza a quelle di primo utilizzo.
3. Manutenzione. Si preferisce mantenere il paziente sotto l’effetto della chemioterapia prima della possibilità di attività leucemica incipiente e che con il trattamento mantiene l’effetto fino a quando la malattia scompare.

Finora per le forme acute, i criteri più severi da considerare CR diventano più complessi, poiché è meglio cercare la remissione molecolare, che comporta la ricerca dell’alterazione cromosomica iniziale come avviene nel caso della leucemia promielocitica (M3-FAB) con t (15;17) iniziale, dovrebbe essere cercato per specifici studi molecolari e citogenetici nonostante sia in CR, poiché se la traslocazione persiste, il trattamento aggressivo deve essere continuato fino alla totale eliminazione del clone. Questa varietà di leucemia dovrebbe essere considerata potenzialmente curabile ed è una delle più comuni nella popolazione latina.
La cura della malattia dipenderà quindi dall’eliminazione di tutte le cellule maligne esistenti nel paziente. In generale, alcune leucemie possono essere suscettibili di guarigione con la sola chemioterapia, ma oggi molta importanza deve essere data ai cosiddetti fattori prognostici basati su modelli matematici che permettono ai pazienti di essere messi in grado di prognosi che hanno e includono:
1. Il tipo di leucemia.
2. L’alterazione molecolare iniziale e la sua persistenza nonostante il trattamento o l’eradicazione.
3. et. I pazienti di età superiore ai 60 anni hanno una prognosi infausta rispetto ai pazienti più giovani.
4. Chemioterapia. Devono essere utilizzati i medicinali indicati e, soprattutto, le dosi raccomandate. Ad esempio, in LAL adulto, l’uso dello schema HyperCyVAD (dosi intensificate di ciclofosfamide, vincristina, adriamicina, desametasone in combinazione con citosina arabinoside e metotrexato) raggiunge il 90% di CR e cure nel 50% dei casi, dati non precedentemente visti con altri schemi. Questo trattamento è tossico e richiede che sia utilizzato solo in istituzioni che dispongono di risorse di supporto sufficienti. Sfortunatamente, nel nostro ambiente non tutti i centri hanno i farmaci raccomandati e i risultati non saranno riproducibili, poiché non hanno abbastanza supporto per il paziente durante la fase di massima mielosoppressione.
5. Terapia di supporto. I risultati della chemioterapia e nuovi farmaci, nuove combinazioni e con più specificità costringere l’attuazione del team multi-disciplinari con l’indirizzo dell’ematologo; installazione e l’uso di cateteri venosi centrali; il supporto per la banca del sangue per il sostegno delle trasfusioni di piastrine ed eritrociti (anche prodotti stellate); l’intervento del infectologist per la rilevazione di infezioni, e l’uso appropriato di antibiotici o agenti antifungini, o profilattiche o terapeutiche, se necessario; la fornitura di sale di isolamento con servizi di manutenzione e quartiermastro che consentono di ottenere un ambiente privo di batteri, compresa l’alimentazione sterile; un laboratorio con protocollo di gestione del campione del paziente in protocolli di trattamento specifici e la gestione di campioni speciali (preparazione di concentrati di leucociti in citometria ematica buffy coat e ottenere una lettura ottimale); di preparazione di farmaci altamente specializzati, e d’altra parte, il più importante è il personale infermieristico, ausiliario e amministrativo, che con l’intero gruppo sono attesi i risultati che hanno in altri paesi.
6. Trapianto di midollo osseo (MO). È un tipo di trattamento complesso e ad alto costo che richiede un donatore MO compatibile e una condizione inattiva con un’alta probabilità di recidiva precoce o tardiva o, con fattori di prognosi infausta. È una procedura con tendenze più curative poiché utilizza megadosi della chemioterapia per sradicare le cellule leucemiche, ma nel tentativo, sradica anche i precursori normali e la sostituzione di un nuovo midollo normale compatibile è necessaria. Il tipo allogenico (fratello compatibile identici) è la scelta migliore dal momento che il ricevitore accetta solo l’identità parziale (l’unica di totale accettazione sono gemelli identici), e, di conseguenza, di un rifiuto della graft-versus-host disease, che a sua volta porta a graft-versus-leukemia e aumenta l’eliminazione di cloni leucemiche, tuttavia, nei casi più gravi, la sindrome (classi 3-4) la morbilità e la mortalità è aumentata nonostante il trattamento specifico.
LEUCEMIE CRONICHE
1. Leucemia linfocitica cronica. Si verifica più frequentemente nelle persone anziane e il criterio è la persistenza della linfocitosi di oltre 10 x 109 / l e MO con infiltrazione di oltre il 50% dei linfociti con fenotipo CD5+. Il criterio del trattamento è la duplicazione di un account di linfociti in un anno o la progressione di adenomegalia o splenomegalia, anche se alcuni casi di là di questo standard per la presenza di anemia emolitica o trombocitopenia autoimmune e quindi indicato il trattamento basato sulla combinazione di fludarabina, ciclofosfamide e prednisone, negli stadi I e II solo somenter e osservati ed attesi evoluzione senza trattamento.
2. Leucemia mieloide cronica (LMC). In questa malattia c’è un grande progresso nella conoscenza della presenza del cromosoma Philadelphia, che è stato descritto nel 1950 nella città dell’Unione Americana, e che il suo inizio è stato il primo cromosoma marcatore in associazione con la malignità, tuttavia nel corso degli anni di ricerca, siamo riusciti a conoscere il t (9;22) con l’espressione funzionale del cromosoma con la produzione di un’oncoproteina con grande attività tirokinochinasi, che aumenta la proliferazione cellulare e che a sua volta spiega la grande leucocitosi e trombocitosi con cui questi pazienti presentano, così come la grande splenomegalia (Figura 5).
In questo momento, sono passati da 10 a 12 anni dalla scoperta di una piccola molecola specificamente diretta contro questo substrato molecolare donatore di fosfato per la regolazione interna della cellula leucemica e dei suoi substrati. Il STI inizialmente chiamato (inibitore di trasduzione del segnale) ha prodotto l’inibizione competitiva della fosforilazione, ha portato all’apoptosi cellulare e i pazienti hanno raggiunto risultati clinici mai visti prima con remissioni molecolari fino all ‘ 80-90% a 10 anni, il che ha cambiato radicalmente la storia naturale della malattia, con il pretrattamento non più di 3 anni. Queste informazioni fanno un cambiamento nella storia naturale e nella prognosi della malattia, precedentemente fatale a breve termine.

Questi esempi sono a base del progresso così intenso che si è verificato negli ultimi anni e che è auspicabile per consentire a suscitare l’interesse degli studenti di medicina, i loro insegnanti, ma soprattutto delle autorità scolastiche della leucemia in generale, che occupa i primi 5 posti della frequenza di neoplasie maligne dell’adulto e i primi posti nei bambini, così ematologia devono essere incluse all’interno del nucleo materie del curriculum di carriera di Medicina e anche come parte di corsi specializzazione post-laurea.
Sebbene utile per la sua semplicità, la classificazione franco-americana britannica (FAB) può portare a errori diagnostici e quindi terapeutici fino al 20% dei casi. Per questo motivo, la classificazione mediante metodi di immunoistochimica e biologia molecolare è diventata una condizione sine qua non per la corretta classificazione e la successiva gestione dei pazienti (Figura 6).

a differenza di classificazione FAB, il CHE (tabella 2) riflette un cambiamento di paradigma attraverso il quale intendiamo malattie del sangue, perché per la prima volta combinato le informazioni genetiche, morfologiche, citochimiche e immunofenotipici con i risultati clinici all’interno di algoritmi diagnostici delle neoplasie del tessuto ematopoietico; l’importanza relativa di ciascun criterio differisce tra le neoplasie e non esiste un “gold standard” per la classificazione di tutte le neoplasie ematologiche. L’obiettivo era definire entità che potessero essere riconosciute dai patologi e che avessero rilevanza clinica. Dalla sua comparsa nel 2001, sono state fatte diverse revisioni per aggiornare il suo contenuto in relazione alle scoperte più recenti. L’ultima revisione risale al 2008 e classifica le neoplasie ematologiche come segue:
1. Neoplasie mieloidi.
2. Neoplasie linfoidi.
3. Mastociti o malattie delle cellule adipose.
4. Malattie delle cellule istiocitiche e dendritiche.

NEOPLASIA MIELOIDE
le neoplasie mieloidi derivano da progenitori nel midollo osseo, si differenziano in eritrociti, granulociti (neutrofili, basofili ed eosinofili), monociti e megacariociti. La classificazione FAB riconosce 3 categorie principali:
1. Leucemia mieloide acuta.
2. Sindromi mielodisplastiche.
3. Neoplasie mieloproliferative.
I determinanti più importanti delle categorie riconosciute utilizzando morfologica, istochimica e immunofenotipica e la percentuale di blasti, il lignaggio cellulare e il grado di differenziazione delle cellule neoplastiche (figura 7). Negli ultimi anni, le caratteristiche genetiche (citogenetica e molecolare), così come il pre-trattamento e l’evoluzione di mielodisplasia, ha mostrato un impatto significativo sul comportamento clinico di queste sofferenze, che non sempre correlano bene con le categorie dei FAB, in modo che il dibattito centrale per la riclassificazione è stata discriminare tra i soggetti patologici e di fattori prognostici, per ottenere una classificazione clinica rilevanza e importanza per il patologo. Alcune anomalie genetiche sembrano definire diverse malattie, mentre altre rappresentano fattori prognostici per una specifica malattia.

Attualmente, la classificazione OMS raggruppa le condizioni mieloidi in 4 gruppi principali:
1. Malattie mieloproliferative
2. Sindromi mielodisplastiche
3. Malattie mielodisplastiche / mieloproliferative
4. Le leucemie mieloidi acute

Le malattie mieloproliferative sono un gruppo di disturbi clonali associati alla proliferazione di una o più linee mieloidi. Sta diventando sempre più chiaro che queste malattie sono spesso associate a mutazioni che causano aumenti anormali dell’attività della tirosina chinasi e della proliferazione delle cellule progenitrici indipendenti dal fattore di crescita nel midollo osseo. La percentuale di blasti nel midollo osseo è normale o leggermente elevata, ma sempre inferiore al 20%. L’emopoiesi è solitamente efficace, con conseguente aumento della conta di una o più cellule mature nel sangue periferico. Il prototipo di neoplasie mieloproliferative è la leucemia mieloide cronica positiva al cromosoma Philadelphia (Ph1) (BCR/ABL). Le altre entità incluse sono:
1. Policitemia vera.
2. Mielofibrosi idiopatica.
3. Trombocitemia essenziale primaria.
4. Leucemia eosinofila cronica.
5. Leucemia neutrofila cronica.
6. Mastocitosi.
7. Neoplasie mieloproliferative inclassificabili.

Le sindromi mielodisplastiche si riferiscono a disturbi caratterizzati da produzione cellulare inefficace e displasia, con un rischio variabile di trasformazione in leucemia acuta. La cellularità nel midollo è spesso aumentata ma altamente variabile. C’è maturazione ma anche displasia di una o più linee mieloidi. L’emopoiesi non è efficace e quindi esistono citopenie. Questo articolo include:
1. Citopenia refrattaria con displasia a una linea*.
• Anemia refrattaria.
• Neutropenia refrattaria.
• Trombocitopenia refrattaria.
2. Anemia refrattaria con sideroblasti anulari.
3. Citopenia refrattaria con displasia di lignaggi multipli.
4. Anemia refrattaria con esplosioni in eccesso.
5. Sindrome mielodisplastica con d (5q).
6. Sindrome mielodispastica inclassificabile.
7. La sindrome mielodisplastica giovanile, include un’entità provvisoria nota come citopenia refrattaria giovanile.
Le sindromi mielodisplastiche / mieloproliferative comprendono disturbi in cui coesistono caratteristiche displastiche e proliferative. Questo gruppo comprende la leucemia giovanile mielomonocitica, che è rappresentativa di entrambe le sindromi (mielodisplastica e mieloproliferativa). Quasi la metà dei pazienti è presente con conta neutrofila normale o bassa e displasia di linee cellulari multiple senza organomegalia e midollo osseo con morfologia simile all’anemia refrattaria con esplosioni in eccesso, ma con monocitosi. Altri pazienti hanno grave neutrofilia, monocitosi e splenomegalia. Non è ancora noto se siano 2 diverse malattie, una mielodisplastica e una mieloproliferativa; tuttavia, finora non ci sono differenze nelle anomalie citogenetiche o nei modelli di crescita delle colonie in vitro o nella loro evoluzione clinica, quindi c’è controversia tra medici e patologi in base al loro posto all’interno della classificazione. Secondo l’ultima revisione, in questa categoria si trovano:
1. Leucemia mielomonocitica cronica.
2. Leucemia mieloide cronica atipica (BCR/ABL negativo).
3. Leucemia mielomonocitica giovanile.
4. Sindrome mielodisplastica / mieloproliferativa non classificabile.
Nella categoria di leucemie acute mieloblásticas (LAM) (che è definito da una percentuale superiore al 20% mieloblasti nel midollo osseo o diluente di sangue periferico, o la presenza di una anomalia citogenetica in particolare, nonostante l’account di blasti) riconosce i seguenti gruppi:
1. LAM con traslocazioni citogenetiche ricorrenti.
2. LAM con caratteristiche mielodisplastiche.
3. LAM e MDS relativi ai trattamenti antineoplastici.
4. Non sono classificabile.
5. Sarcoma mieloide.
6. Proliferazioni mieloidi correlate alla sindrome di Down.
7. Neoplasia blastica plasmacitoide di cellule dendritiche.
Le NEOPLASIE LINFOIDI
sono quelle che provengono da cellule che normalmente si sviluppano in linfociti T (LT citotossici, collaboratori o regolatori) o linfociti B (linfociti o plasmacellule). In generale, le neoplasie linfoidi si dividono in quelle derivate da precursori linfoidi e quelle da linfociti maturi e plasmacellule e vengono successivamente raggruppate in base al loro lignaggio (B o T).
Storicamente, le neoplasie linfoidi che si verificano nel midollo osseo e coinvolgono il midollo osseo sono state separate da quelle che si presentano come tumore (linfoma). Tuttavia, è ora noto che qualsiasi linfoma può presentare caratteristiche cliniche della leucemia e che qualsiasi leucemia può occasionalmente presentarsi come tumore (sarcoma granulocitico). Nella classificazione OMS, la diagnosi di diverse neoplasie linfoidi dipende non solo dalla posizione anatomica delle cellule tumorali, ma anche dall’origine morfologicamente definita della cellula tumorale. Queste considerazioni hanno annullato la rilevanza dei termini L1 e L2 della classificazione FAB, poiché non sono correlati con il loro immunofenotipo, anomalie genetiche o con il loro decorso clinico (Figura 8). L3 è equivalente al linfoma di Burkitt nella fase leucemica e dovrebbe essere diagnosticato come tale.

1. Neoplasie precursori. Esiste un consenso sul fatto che le neoplasie precursori che si presentano come tumori solidi e quelle che coinvolgono il midollo osseo e il sangue sono biologicamente la stessa malattia con diverse presentazioni cliniche. La maggior parte delle neoplasie linfoidi precursori sono presentate come leucemie, quindi è stato convenuto che la classificazione dovrebbe mantenere il termine LAL per la fase leucemica delle neoplasie precursori di tipo B e T. Esistono 2 categorie principali:
* Leucemie / linfomi precursori B.
• Leucemie / linfomi precursori T.
2. Neoplasie a cellule B mature. La classificazione proposta considera linfomi e leucemie dello stesso tipo cellulare della stessa malattia con diverse presentazioni o fasi cliniche. Le malattie specifiche derivate dalle cellule B mature sono le seguenti:
1. Leucemia linfocitica cronica / linfoma linfocitario piccolo.
2. Linfoma linfoplasmacitico.
3. Linfoma mantellare.
4. Leucemia a cellule B prolinfocitica.
5. Linfoma follicolare.
6. Linfoma diffuso a grandi cellule B.
* Linfoma intravascolare a grandi cellule B.
• Linfoma primario mediastinico a grandi cellule B.
• Linfoma a grandi cellule B (correlato al virus di Epstein Barr-EBV).
•• Linfoma a grandi cellule B ricco di istiociti e cellule T.
* Sistema nervoso centrale diffuso linfoma a grandi cellule B.
• * Linfoma cutaneo primario diffuso a grandi cellule B.
• Linfoma diffuso a grandi cellule B degli anziani positivo per EBV.
• Linfoma pasmablastico.
• Linfoma pleurico primario.
• * Alkoma-positivo (ALK) linfoma a grandi cellule.
• Linfoma di Burkitt.
7. Zona marginale Linfoma a cellule B.
8. Linfoma a cellule B della zona marginale extranodale.
9. Zona marginale splenica Linfoma a cellule B
10. Leucemia a cellule capellute.
11. Plasmocitoma / mieloma plasmacellulare.

NEOPLASIE DELLA LINEA MIELOIDE E LINFOIDE
Alcune neoplasie esprimono marcatori di entrambe le linee mieloide e linfoide.indifferenziata) o presenti caratteristiche di entrambe le linee (fenotipo misto o linea mista leucemia acuta).
Tabella 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍA
Bassan R, Hoelzer D. Terapia moderna della leucemia linfoblastica acuta. J Clin Oncol. 2011;29:523-43.
Burnett A, Wetzler M, Löwenberg B. Progressi terapeutici nella leucemia acuta. J Clin Oncol. 2011;29:487-94.
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. La classificazione OMS 2008 delle neoplasie linfoidi e oltre: concetti in evoluzione e applicazioni pratiche. Sangue. 2011 Maggio 12; 117 (19): 5019-32. Epub 2011 Febbraio 7. Recensione.
Cortes J, Hochhaus A, Hugues T, Kantajian H. Terapie di prima linea e di salvataggio con inibitori della tirosina chinasi e altri trattamenti nella leucemia mieloide cronica. J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologia, stratificazione del rischio e terapia della leucemia acuta pediatrica: un aggiornamento. J Clin Oncol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado MR. Valutazione multicentrica indipendente di ourcomes in pazienti affetti da leucemia mieloide cronica trattati con Imatinib. Rivista National Cancer Institute. 2011;103:1-9.
Grever M, Lozanki G. Strategie moderne per la leucemia a cellule capellute. J Clin Oncol. 2011:29;583-90.
Gribben JG, O’Brien S. Aggiornamento sulla terapia della leucemia linfocitica cronica. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Leucemia mieloide cronica. Concetti attuali in Fisiopatologia e Trattamento. Cancerología. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib confrontato con Imatinib / Citarabina per il trattamento di prima linea della leucemia mieloide cronica positiva al cromosoma Philadelphia precoce. Risultati di uno studio clinico randomizzato del gruppo messicano di leucemia collaborativa. Leucemia clinica. 2008: 2(2);1128-32.
Lichtman MA. Classificazione e manifestazioni cliniche dei disturbi mieloidi clonali. It: Williams. Ematologia. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Genetica molecolare della leucemia mieloide acuta adulta. Implicazioni prognostiche e terapeutiche. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiologia della Citometria Ematica. Rev. Fac Med UNAM. 2010; 53:36-43.
Sanz M, Lo-Coco F. Approcci moderni al trattamento della leucemia promielocitica acuta. J Clin Oncol. 2011;29:495-503.
Nota
* Displasia. Si riferisce all’alterazione citomorfologica che include la dissociazione della maturazione nucleo-citoplasma (ricorda che la maturazione della cromatina dipende dal citoplasma del DNA e dell’RNA, quindi il nucleo si ferma alla maturazione mentre il citoplasma continua il suo normale processo) che produce cellule non vitali e c’è apoptosi intramidollare.