Un maschio di 49 anni con una storia di abuso di alcol presenta all’ED lamentele di dolore addominale generalizzato e vomito per le ultime 36 ore. Il paziente è ben noto al dipartimento per le visite legate all’alcol e continua a bere ogni giorno. All’arrivo, è tachicardico e tachipneico, e i risultati degli esami fisici includono mucose secche, diminuzione del turgore di sakin, tenerezza epigastrica e tremore in entrambe le mani. Studi di laboratorio mostrano un bicarbonato sierico di 10 mEq / L, un gap anionico di 30, un glucosio sierico di 95 mg/dL, un’acidosi lattica con pH 7,2, ipofosfatemia e chetonuria di tracce. La TAC addominale è normale. Nega una storia di diabete mellito, ingestione di alcoli tossici o malattia recente.
Questo paziente potrebbe potenzialmente avere una delle molte diagnosi, ma la sua presentazione e i risultati di laboratorio sono più coerenti con la chetoacidosi alcolica (AKA). AKA può essere una diagnosi ED comune e si verifica in genere nei bevitori di alcol cronici che hanno una brusca cessazione nella loro assunzione di alcol accoppiato con una diminuzione dell’assunzione glicemica e l’esaurimento del volume intravascolare.1
Nella maggior parte dei casi, un evento precipitante come pancreatite, gastrite o polmonite da aspirazione porta ad un brusco calo dell’assunzione orale. Circa 24 a 72 ore dopo la cessazione dell’assunzione di PO, AKA può svilupparsi.2 Questi pazienti di solito hanno una bassa o assente concentrazione sierica di alcol e possono presentare con vari gradi di astinenza da alcol. Tuttavia, un sensorio chiaro è un segno distintivo di questa condizione. La presenza di un’alterazione della coscienza suggerisce fortemente che è presente un altro processo.3
Sebbene la fisiopatologia sottostante sia complessa, una corretta comprensione aiuta notevolmente nella diagnosi e nella gestione di questa condizione.
Ci sono tre concetti generali che guidano AKA:
- L’ingestione di alcol, aggravata da una diminuzione dell’apporto calorico e dalla disidratazione, favorisce uno stato chetotico.
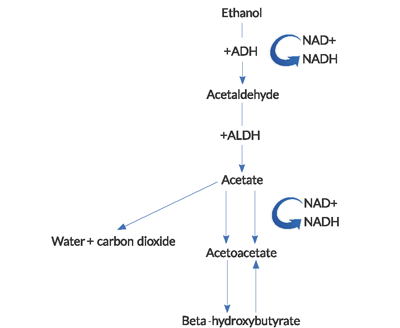
La chetoacidosi è causata da una combinazione di fattori, tra cui ipoinsulinemia indotta da fame, ossidazione dell’alcol ai suoi vari metaboliti chetonici, lipolisi con rilascio di acido grasso libero (FFA) e contrazione del volume intravascolare. Lo stato di fame relativo in AKA conduce all’eccessiva secrezione del glucagone ed alle concentrazioni periferiche ridotte dell’insulina, che svolge un ruolo chiave nello sviluppo della chetoacidosi. Il metabolismo dei grassi attraverso la lipolisi produce beta-idrossibutirrato (BHB) e acitil-acetato (ACA). Questi chetoni sono utilizzati per la respirazione cellulare per fornire energia attraverso la produzione di adenosina trifosfato (ATP), ma aggiungono all’acidosi anionica osservata in AKA.
- Durante il metabolismo dell’etanolo, vengono generate elevate quantità di NADH (la forma ridotta di nicotinamide-adenina dinucleotide).4
NAD+ è un coenzima utilizzato per trasportare elettroni nelle reazioni redox intracellulari. La riduzione di NAD+ e conseguente accumulo e squilibrio di NADH nel metabolismo dell’etanolo ha diverse conseguenze importanti. La generazione di BHB predomina sulla produzione di ACA in questo alto rapporto NADH / NAD+. Questo rapporto anormale porta ad un’inibizione del ciclo dell’acido citrico e della gluconeogenesi epatica, che spiega parzialmente perché l’iperglicemia è rara in questi pazienti.Quasi controintuitivamente, c’è un fallimento nel rigenerare i livelli normali di NAD+ e ACA in AKA. La reossidazione di NADH a NAD+ sembra essere limitata da una combinazione di fattori, tra cui l’ipofosfatemia e un blocco funzionale all’interno dei mitocondri.2L’acidosi lattica osservata in AKA è dovuta a uno stato redox anormale. Il piruvato è un substrato utilizzato in numerose vie di produzione di energia, ma nella chetoacidosi alcolica, viene spostato dalle sue normali vie metaboliche ad altre che aumentano la produzione di lattato. Inoltre, la rigenerazione del piruvato dall’acido lattico è compromessa.
- Uno stato adrenergico accentuato e l’esaurimento del volume peggiorano la chetosi e inibiscono la gluconeogenesi, creando uno stato che favorisce la creazione e il mantenimento di un ambiente chetotico.
Il corpo risponde alla fame, alla disidratazione e all’ipoglicemia con il rilascio di ormoni controregolatori. Questi ormoni aumentano il tono simpatico, diminuiscono il rilascio di insulina e aumentano la concentrazione di chetoni attraverso il rilascio di FFA e diminuisce il metabolismo dei chetoni periferici. Tutti questi cambiamenti perpetuano lo stato chetotico fino a quando il glucosio non viene reintrodotto nel sistema. La disidratazione significativa dovuta al vomito e alla diminuzione dell’assunzione orale portano a una compromissione della clearance dei chetoni renali, esacerbando ulteriormente la situazione.2La diagnosi differenziale per AKA dovrebbe includere chetosi da fame e chetoacidosi diabetica (DKA). Anche se una storia approfondita può aiutare a restringere il differenziale, un pannello metabolico è essenziale per confermare la diagnosi. Lacune anioniche di 30 mEq / L o più può essere visto in AKA, anche se il divario può essere oscurato da una concomitante alcalosi metabolica primaria a causa di vomito. Infatti, ci sono casi di pazienti con AKA che hanno un pH sierico alcalemico a causa di vomito eccessivo.Il divario anionico nella chetosi da fame è tipicamente molto più basso, con livelli di bicarbonato raramente inferiori a 18 mEq/L e pH sierico tipicamente superiore a 7,30.2 In DKA, al contrario, il divario anionico può essere piuttosto elevato, con livelli di bicarbonato che raggiungono frequentemente le singole cifre. L’iperglicemia con glicosuria, tipicamente osservata nella chetoacidosi diabetica (DKA), è rara con AKA.4 La malnutrizione cronica porta a basse riserve di glicogeno e l’aumento del tono adrenergico porta all’inibizione della gluconeogenesi epatica. Chetonuria, presente in tutte e tre queste condizioni, può confondere la gravità di AKA.La chetonuria viene misurata dal test nitroprussiato, in cui un cambiamento di colore indica la concentrazione relativa di acetone e ACA nelle urine. La presenza di BHB, il chetone più importante presente in AKA, non viene riflessa dal test del nitroprussiato. Questo spiega perché i pazienti con AKA possono mostrare nessuna o solo lieve chetonuria sulla presentazione iniziale, con un aumento paradossale come la condizione è invertita. Man mano che il rapporto ACA:BHB si normalizza, sia l’ACA rilevabile che il BHB vengono eliminati nelle urine.
Diagnosi differenziale
Nella diagnosi differenziale devono essere prese in considerazione anche altre condizioni potenzialmente letali che possono causare una significativa acidosi da gap anionico. Gli alcoli tossici, in particolare metanolo e glicole etilenico, possono essere ingeriti intenzionalmente o accidentalmente in questa popolazione di pazienti. Queste ingestioni possono causare morbilità e mortalità significative se non gestite in modo appropriato.5 Stato mentale alterato è una caratteristica comune di ingestione di alcol tossico, ma di solito non è visto in AKA.5
I pazienti avranno tipicamente una lacuna osmolare iniziale che le transizioni ad una lacuna aumentata dell’anione mentre l’alcool tossico è metabolizzato. Elevata concentrazione di BHB nel siero può essere abbastanza elevata in AKA, ma questo non esclude necessariamente la possibilità di ingestione di alcol tossico; né l’assenza di un gap osmolare o anionico esclude la diagnosi. Mentre i pazienti in AKA hanno una leggera acidosi lattica, la presenza di un livello di lattato significativamente elevato dovrebbe indurre la ricerca di una malattia di base. Raramente, una combinazione di AKA e uno di questi altri eventi può verificarsi e presentare un enigma diagnostico. Una considerazione ponderata dei tempi, del tipo e della quantità di ingestione e dei sintomi associati, in combinazione con studi di osservazione e di laboratorio, deve essere utilizzata per effettuare questa differenziazione se manca una storia chiara e accurata.
Trattamento
L’inversione della chetosi e la reidratazione vigorosa sono centrali nella gestione dell’AKA. Oltre alla sostituzione del fluido isotonico, sono necessari fluidi endovenosi contenenti destrosio. Tipicamente, il 5% di destrosio con soluzione salina semis normale ad una velocità di 150 ml all’ora fornisce glucosio sufficiente per stimolare il pancreas a secernere insulina, consentendo ai tessuti periferici di metabolizzare i chetoni e inibire il rilascio di FFA.2 Permette anche al corpo di rigenerare NAD+, che è inibito dalle alterazioni metaboliche causate da AKA. Le infusioni di liquidi contenenti destrosio per via endovenosa devono essere interrotte una volta che i livelli di bicarbonato hanno raggiunto 18-20 mEq/L e il paziente sta tollerando l’assunzione orale. Ciò si verifica in genere da 8 a 16 ore dopo l’inizio del trattamento.2 L ‘ astinenza da alcol in questi pazienti deve essere gestita in modo aggressivo con benzodiazepine per via endovenosa. Tiamina, folato e altri elettroliti, in particolare fosfato e potassio, possono dover essere reimpiegati in questi pazienti.6 È interessante notare che la maggior parte della morbilità osservata in AKA è dovuta al processo sottostante che ha causato la cessazione dell’alcol.
Conclusione del caso
Il paziente ha ricevuto 4 litri di soluzione salina normale ed è stato avviato su D5-1/2 NS prima del ricovero. Gli è stato somministrato IV valium per astinenza da alcol, e tiamina, folato e fosfato sono stati repletati. E ‘ stato ricoverato in ospedale per tre giorni per la gestione di AKA e ritiro di alcol, poi dimesso una volta tollerando l’assunzione orale e in buone condizioni. È stato visto tre settimane dopo nel dipartimento di emergenza per una presentazione simile.
Tabella 1. Characteristics of Common Ketoacidoses
| Diabetic Ketoacidosis | Alcoholic Ketoacidosis | Starvation Ketoacidosis | |
| Bicarbonate | Can reach single digits | Can reach single digits | > 18 |
| Glucose | Elevated | Low to mildly elevated | Low to normal |
| Measurable ketonuria | Present | Absent or present | Present |
Figure 1. Pathway of alcohol metabolism
(ADH = alcol deidrogenasi, ALDH = acetaldeide deidrogenasi).
- Palmer, Jerry P. Chetoacidosi alcolica: presentazione clinica e di laboratorio, fisiopatologia e trattamento. Cliniche in endocrinologia e metabolismo 12.2 (1983): 381-389.
- Duffens K, Marx JA. Recensione di ketoacidosisa alcolica.Il Journal of emergency medicine 5.5 (1987): 399-406.
- Wrenn KD, Slovis CM, Minion GE, et al. La sindrome della chetoacidosi alcolica. L’American journal of medicine 91.2 (1991): 119-128.
- Marx JA, Hockberger RS, Walls RM, et al., eds. Rosens™ Medicina di emergenza: concetti e pratica clinica. Filadelfia, PA: Mosby / Elsevier; 2013. Capitolo 185 Malattia correlata all’alcol di John T. Finnell.
- Kraut JA, Kurtz I. â € œToxic alcol ingestions: caratteristiche cliniche, diagnosi, e la gestione. Clinical Journal della Società Americana di Nefrologia 3.1 (2008): 208-225.
- Miller PD, Heinig R, Waterhouse C. Trattamento dell’acidosi alcolica: il ruolo del destrosio e del fosforo. Archives of internal medicine 138.1 (1978): 67-72.