Article de revue
Leucémie – pour le médecin généraliste
Leucémie pour le médecin généraliste
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
Chef du département d’hématologie. Hôpital ÁnGeles del Pedregal. Mexique, DF. Adresse e-mail: [email protected]
b Médecine interne. Hôpital ÁnGeles del Pedregal. Mexique. DF.
c Médecine interne. Hôpital ÁnGeles del Pedregal. Mexique. DF.
Reçu: 17 octobre 2011
Accepté: 07 Janvier 2012
INTRODUCTION
Malgré les grandes avancées moléculaires et thérapeutiques dans l’étude des leucémies, les aspects fondamentaux de cette affection ne sont pas encore clairement connus par le non-hématologue, l’objectif de ce travail est donc de fournir des informations fondamentales aux étudiants en médecine et aux médecins en général, et cela permet surtout d’acquérir des connaissances générales sur les leucémies, leur diagnostic rapide et de rechercher rapidement une référence auprès de l’hématologue.

DÉFINITION
La leucémie est le terme utilisé pour définir un groupe de maladies du sang malignes. Un diagnostic précoce est essentiel, car il permettra au patient d’aller tôt avec le spécialiste en hématologie, qui dirigera le processus de diagnostic et proposera le traitement spécifique. Elle se caractérise par une prolifération clonale, autonome et anormale des cellules qui donnent naissance au reste des cellules normales du sang (comportement tumoral en général).
Cela implique qu’une cellule précoce subit un changement génétique qui provoquera un clone anormal (colonie) d’elle-même de manière incontrôlable. Cette production anormale est désordonnée car les cellules anormales se multiplient à l’image et à la ressemblance d’elles-mêmes, de sorte qu’elles occupent progressivement l’espace de la moelle osseuse normale et provoquent une anémie progressive, des saignements anormaux et une prédisposition aux infections. D’autre part, lorsque des cellules anormales envahissent d’autres tissus, il y aura une défaillance du fonctionnement de l’organe concerné, par exemple, une infiltration dans le système nerveux central qui se produit dans la leucémie lymphoblastique aiguë (LAL) pourrait se manifester par des maux de tête, des convulsions, des altérations motrices focalisées, une augmentation de la pression intracrânienne, et l’incapacité de poser un diagnostic précoce et de fournir un traitement adéquat, entraînera une perte de fonction et des conséquences irréversibles.
MANIFESTATIONS CLINIQUES
Le tableau clinique est varié et dépend du type de leucémie: aiguë ou chronique, cependant pour les 2 il existe des manifestations cliniques non spécifiques (qui surviennent dans n’importe quelle maladie):
1. Fatigue.
2. Fatigue facile.
3. Faiblesse généralisée.
4. Désire rester au repos ou au lit.
5. Cela nécessite l’aide de quelqu’un pour répondre à vos besoins personnels.
Les leucémies chroniques sont indolentes et jusqu’à 50% des cas sont découverts lors d’une revue clinique ou de laboratoire de routine chez des volontaires considérés comme en bonne santé et venus donner du sang, cependant, à mesure que la maladie progresse, des manifestations non spécifiques sont présentées mais maintenant elles sont spécifiques (tableau 1).

Dans les formes aiguës, des manifestations spécifiques sont dérivées d’une déficience de l’une des lignées cellulaires:
1. Érythrocytes: syndrome anémique dont l’intensité dépendra du degré d’hypoxémie quel que soit le degré d’anémie. Dyspnée d’effort moyen jusqu’à orthoprea.
2. Plaquettes: pétéchies, ecchymose des extrémités et, dans les cas généralisés plus sévères, hémorragie sèche et humide avec épistaxis, gingivorragie, hématurie, crinière ou hématochésie. Très grave dans le système nerveux central (SNC).
3. Leucocytes: fièvre, diaphorèse, infections localisées jusqu’à une septicémie franche (bactéries ou champignons). Ils surviennent avec une neutropénie inférieure à 250 neutrophiles totaux / mm3.
Syndrome infiltrant: se réfère à une implantation anormale dans n’importe quel tissu, bien qu’elle soit fréquente:
1. Hépatomégalie ou splénomégalie (Figure 5).
2. Adénomégalie (locale ou généralisée).
3. Teint leucémique.
4. Douleur osseuse due à l’expansion de la moelle osseuse.
5. Tissus mous (sarcome granulocytaire).
6. Testicule.
7. SNC.
8. Gencives et tout site (figure 1).

Troubles métaboliques: ils résultent d’une hyperproduction anormale de cellules malignes et d’une apoptose accrue.
1. Acidose.
2. Augmentation de la déshydrogénase lactique (LLD).
3. Hyperkaliémie.
4. Hyperuricémie.
5. Augmentation de la microglobuline β2.

La preuve clinique prédomine comme la pierre angulaire des diagnostics suspects de leucémies aiguës et de toute affection, mais ce qui suit est de compléter le diagnostic avec le soutien du laboratoire clinique dans la cytométrie numération formule sanguine complète, ou spéciale, c’est-à-dire l’observation approfondie du frottis de sang périphérique par le personnel technique qui a la préparation dans l’identification des cellules anormales, en particulier la leucémie.

Les modifications de laboratoire nécessitant un examen spécial comprennent :
1. Anémie (tout grade).
2. Leucopénie ou leucocytose (prédominance d’une lignée cellulaire).
3. Thrombocytopénie.
4. Combinaisons: bicitopénie ou pancytopénie.
Des précautions particulières doivent être prises lorsque le laboratoire signale la présence de leucocytes ou de lymphocytes atypiques (pouvant être des blastes leucémiques). Il est conseillé de demander un examen par un expert (figure 2).

L’aspiration de la moelle osseuse est essentielle au diagnostic (figure 3) et 20% des blastes sont nécessaires pour établir les critères de leucémie aiguë dans l’une de ses variétés.

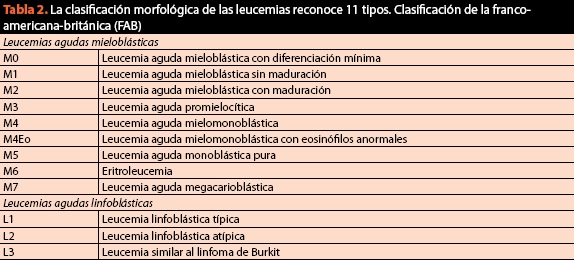
Dans la même procédure, des échantillons doivent être obtenus pour la classification finale de l’affection et demander le caryotype et l’immunophénotype, car actuellement le critère cytomorphologique est d’une importance vitale mais n’est plus suffisant. La classification actuelle et actuelle des maladies du sang malignes est répertoriée dans le tableau 2.
Le traitement vise 2 aspects importants: le premier d’entre eux est l’antileucémique spécifique et repose sur l’utilisation de médicaments d’origine chimique appelés chimiothérapie, dont l’objectif principal est d’éradiquer, c’est-à-dire d’éliminer toutes les cellules leucémiques du corps. Le deuxième aspect du traitement est le soutien des complications qui surviennent généralement chez les patients à l’admission.
Le traitement vise 2 aspects importants: le premier d’entre eux est l’antileucémique spécifique et repose sur l’utilisation de médicaments d’origine chimique connus sous le nom de chimiothérapie, dont l’objectif principal est d’éradiquer, c’est-à-dire d’éliminer toutes les cellules leucémiques du corps.
Le deuxième aspect du traitement est le soutien aux complications que les patients présentent habituellement à l’admission, telles que:
1. Anémie.
2. Saignement anormal.
3. Infections pulmonaires et généralisées, entre autres (figure 4).
4. Toute autre complication adjacente que le patient peut avoir (comorbidité), telle que des conditions préexistantes, par exemple le diabète, l’hypertension, les maladies cardiaques et d’autres maladies courantes chez les patients atteints de leucémie.


par conséquent, il est très important de prendre en compte que le traitement contre la leucémie est multidisciplinaire, impliquant la participation d’autres spécialistes tels que le soutien à l’hématologue.
Le traitement antileucémique sera également différent pour différents types de leucémie et pour les formes aiguës. Il est divisé en 3 phases :
1. Induction de rémission. L’objectif est d’obtenir une rémission complète (CR), c’est-à-dire une normalisation des valeurs sanguines du patient, l’absence de symptômes ou de signes indiquant que la leucémie persiste avec infiltration. Au cours du processus, le patient devrait avoir un « état sans leucémie » dans la moelle osseuse et l’avenir devrait être la récupération à une hématopoïèse normale, et malheureusement dans d’autres cas, ils se rétablissent avec la maladie, qui parle de leucémie résistante dont le pronostic est terrible. Ce premier processus peut prendre de 6 à 8 semaines pour atteindre la CR.
2. Consolidation. Il implique l’utilisation des mêmes médicaments qui ont été utilisés dans l’induction ou la combinaison d’autres chimiothérapeutiques, également dans le but de suivre l’éradication des cellules malignes résiduelles qui pourraient développer une résistance aux cellules de première utilisation.
3. Entretien. Il est préférable de garder le patient sous l’effet de la chimiothérapie avant la possibilité d’une activité leucémique naissante et qu’avec le traitement, il maintienne son effet jusqu’à ce que la maladie disparaisse.

Jusqu’à présent pour les formes aiguës, les critères les plus stricts à considérer CR deviennent plus complexes, car il est préférable de rechercher une rémission moléculaire, ce qui implique la recherche de l’altération chromosomique initiale comme cela se produit dans le cas de la leucémie promyélocytaire (M3-FAB) avec t (15;17) initialement, il convient de rechercher des études moléculaires et cytogénétiques spécifiques malgré le fait d’être en CR, car si la translocation persiste, le traitement agressif doit être poursuivi jusqu’à l’élimination totale du clone. Cette variété de leucémie doit être considérée comme potentiellement curable et est l’une des plus courantes dans la population latino-américaine.
La guérison de la maladie dépendra alors de l’élimination de toutes les cellules malignes existantes chez le patient. En général, certaines leucémies peuvent être susceptibles de guérir par la chimiothérapie seule, mais il faut aujourd’hui accorder une grande importance aux facteurs dits pronostiques qui sont basés sur des modèles mathématiques qui permettent de placer les patients dans le degré de pronostic qu’ils ont et comprennent:
1. Le type de leucémie.
2. L’altération moléculaire initiale et sa persistance malgré le traitement ou l’éradication.
3. âge. Les patients de plus de 60 ans ont un mauvais pronostic par rapport aux patients plus jeunes.
4. Chimiothérapie. Les médicaments indiqués et, surtout, les doses recommandées doivent être utilisés. Par exemple, chez l’adulte LAL, l’utilisation du schéma HyperCyVAD (doses augmentées de cyclophosphamide, de vincristine, d’adriamycine, de dexaméthasone en association avec l’arabinoside de cytosine et le méthotrexate) atteint 90% de CR et guérit dans 50% des cas, données jamais vues auparavant avec d’autres schémas. Ce traitement est toxique et nécessite qu’il ne soit utilisé que dans des institutions disposant de ressources de soutien suffisantes. Malheureusement, dans notre environnement, tous les centres n’ont pas les médicaments recommandés et les résultats ne seront pas reproductibles, car ils n’ont pas assez de soutien pour le patient pendant la phase de myélosuppression maximale.
5. Thérapie de soutien. Les réalisations de la chimiothérapie et de nouveaux médicaments, de nouvelles combinaisons et avec plus de spécificité obligent la mise en place des équipes pluridisciplinaires à l’adresse de l’hématologue; installation et utilisation de cathéters veineux centraux; soutien à la banque de sang pour le soutien des transfusions de plaquettes et d’érythrocytes (même des produits étoilés); l’intervention de l’infectiologue pour la détection des infections, et l’utilisation appropriée d’antibiotiques ou d’agents antifongiques, prophylactiques ou thérapeutiques, si nécessaire; la mise à disposition de salles d’isolement avec des services d’entretien et d’intendance qui permettent d’atteindre un environnement exempt de bactéries, y compris une alimentation stérile; un laboratoire avec protocole de gestion des échantillons patients dans des protocoles de traitement spécifiques et la gestion d’échantillons spéciaux (préparation de concentrés de leucocytes en cytométrie hématique buffy coat et obtention d’une lecture optimale); domaine de préparation de médicaments hautement spécialisés, et d’autre part, le plus important est le personnel infirmier, auxiliaire et administratif, qui, avec l’ensemble du groupe, sont attendus des résultats qu’ils ont dans d’autres pays.
6. Greffe de moelle osseuse (MO). C’est un type de traitement complexe et coûteux qui nécessite un donneur de MO compatible et une condition inactive avec une forte probabilité de rechute précoce ou tardive ou, avec des facteurs de mauvais pronostic. C’est une procédure avec des tendances plus curatives car elle utilise des mégadoses de chimiothérapie pour éradiquer les cellules leucémiques, mais dans la tentative, elle éradique également les précurseurs normaux et le remplacement d’une nouvelle moelle normale compatible est nécessaire. Le type allogénique (compatible fraternel identique) est la meilleure sélection puisque le récepteur n’accepte que par identité partielle (les seuls d’acceptation totale sont des jumeaux identiques), et se traduit donc par un rejet de la maladie du greffon contre l’hôte, ce qui entraîne à son tour une leucémie du greffon contre l’hôte et augmente l’éradication des clones leucémiques, cependant, dans les cas graves du syndrome (grades 3-4), la morbidité et la mortalité sont augmentées malgré la manipulation spécifique.
LEUCÉMIES CHRONIQUES
1. Leucémie lymphocytaire chronique. Elle survient plus fréquemment chez les personnes âgées et le critère est la persistance d’une lymphocytose supérieure à 10 x 109 / l, et MO avec infiltration de plus de 50% des lymphocytes de phénotype CD5+. Le critère de traitement est la duplication du compte des lymphocytes en une année ou la progression de l’adénomégalie ou de la splénomégalie, bien que certains cas dépassent cette norme par la présence d’anémie hémolytique ou de thrombocytopénie, auto-immune, puis indiquent le traitement basé sur l’association de la fludarabine, du cyclophosphamide et de la prednisone, aux stades I et II seulement somenter et évolution observée et attendue sans traitement.
2. Leucémie myéloïde chronique (LMC). Dans cette maladie, il y a un grand progrès dans la connaissance de la présence du chromosome de Philadelphie, qui a été décrit en 1950 dans la ville de l’Union américaine, et que sa création était le premier chromosome marqueur en association avec la malignité, cependant au fil des années de recherche, nous avons réussi à connaître le t (9;22) avec l’expression fonctionnelle du chromosome avec la production d’une oncoprotéine à grande activité thyrokinokinase, qui augmente la prolifération cellulaire et qui à son tour explique la grande leucocytose et la thrombocytose avec lesquelles ces patients se présentent, ainsi que la grande splénomégalie (Figure 5).
A cette époque, 10 à 12 ans se sont écoulés depuis la découverte d’une petite molécule spécifiquement dirigée contre ce substrat moléculaire donneur de phosphate pour la régulation interne de la cellule leucémique et de ses substrats. L’inhibiteur de transduction du signal initialement appelé IST (inhibiteur de transduction du signal) a produit une inhibition compétitive de la phosphorylation, a conduit à une apoptose cellulaire et les patients ont obtenu des résultats cliniques jamais vus auparavant avec des rémissions moléculaires allant jusqu’à 80-90% à 10 ans, ce qui a radicalement changé l’histoire naturelle de la maladie, le prétraitement ne dépassant pas 3 ans. Ces informations modifient l’histoire naturelle et le pronostic de la maladie, auparavant mortelle à court terme.

Ces exemples sont une base des progrès si intenses qui se sont produits ces dernières années et qu’il est souhaitable de permettre de susciter l’intérêt des étudiants en médecine, de leurs enseignants, mais surtout des autorités éducatives de la leucémie en général, qui occupe les 5 premières places de la fréquence de la maladie maligne de l’adulte et des premières places chez les enfants, de sorte que l’hématologie sera incluse dans les matières de base du programme de la carrière de la Médecine et même dans le cadre des cours spécialisation de troisième cycle.
Bien qu’utile pour sa simplicité, la classification franco-américaine britannique (FAB) peut conduire à des erreurs diagnostiques et donc thérapeutiques dans jusqu’à 20% des cas. Pour cette raison, la classification par les méthodes d’immunohistochimie et de biologie moléculaire est devenue une condition sine qua non pour la classification correcte et la prise en charge ultérieure des patients (figure 6).

contrairement à la classification FAB, l’OMS (tableau 2) reflète un changement de paradigme à travers lequel nous comprenons les maladies du sang, car pour la première fois combiné les informations génétiques, morphologiques, cytochimiques et immunophénotypiques avec les résultats cliniques au sein des algorithmes de diagnostic des néoplasmes du tissu hématopoïétique; l’importance relative de chaque critère diffère selon les néoplasmes et il n’existe pas de « étalon-or » pour la classification de toutes les tumeurs malignes hématologiques. L’objectif était de définir des entités pouvant être reconnues par les pathologistes et présentant une pertinence clinique. Depuis son apparition en 2001, différentes révisions ont été apportées pour mettre à jour son contenu par rapport aux découvertes les plus récentes. Le dernier examen datait de 2008 et classait les tumeurs malignes hématologiques comme suit:
1. Néoplasmes myéloïdes.
2. Néoplasmes lymphoïdes.
3. Maladies des mastocytes ou des cellules adipeuses.
4. Maladies des cellules histiocytaires et dendritiques.

NÉOPLASIES MYÉLOÏDES
les néoplasmes myéloïdes sont dérivés de progéniteurs dans la moelle osseuse, se différencient en érythrocytes, granulocytes (neutrophiles, basophiles et éosinophiles), monocytes et mégacaryocytes. La classification FAB reconnaît 3 catégories principales :
1. Leucémie myéloïde aiguë.
2. Syndromes myélodysplasiques.
3. Néoplasmes myéloprolifératifs.
Les déterminants les plus importants des catégories reconnues à l’aide de la morphologie, de l’histochimie et de l’immunophénotypie et du pourcentage de cellules blastiques, de la lignée cellulaire et du degré de différenciation des cellules néoplasiques (figure 7). Ces dernières années, les caractéristiques génétiques (cytogénétiques et moléculaires), ainsi que le prétraitement et l’évolution de la myélodysplasie, ont montré un impact significatif sur le comportement clinique de ces souffrances, qui ne sont pas toujours bien corrélées avec les catégories de la FAB, de sorte que le débat central pour la reclassification était de discriminer entre les entités et les facteurs pronostiques pathologiques, pour obtenir une classification avec pertinence clinique et signification pour le pathologiste. Certaines anomalies génétiques semblent définir différentes maladies, tandis que d’autres représentent des facteurs pronostiques pour une maladie spécifique.

Actuellement, la classification de l’OMS regroupe les affections myéloïdes en 4 groupes principaux:
1. Maladies myéloprolifératives
2. Syndromes myélodysplasiques
3. Maladies myélodysplasiques / myéloprolifératives
4. Les leucémies myéloïdes aiguës

Les maladies myéloprolifératives sont un groupe de troubles clonaux associés à la prolifération d’une ou plusieurs lignées myéloïdes. Il devient de plus en plus clair que ces maladies sont souvent associées à des mutations qui provoquent une augmentation anormale de l’activité de la tyrosine kinase et une prolifération cellulaire progénitrice indépendante du facteur de croissance dans la moelle osseuse. Le pourcentage de blastes dans la moelle osseuse est normal ou légèrement élevé, mais toujours inférieur à 20%. L’hématopoïèse est généralement efficace, ce qui entraîne une augmentation du nombre d’une ou plusieurs cellules matures dans le sang périphérique. Le prototype des néoplasmes myéloprolifératifs est la leucémie myéloïde chronique (BCR/ABL) à chromosome Philadelphie positif (Ph1). Les autres entités incluses sont :
1. Polycythémie vera.
2. Myélofibrose idiopathique.
3. Thrombocytémie essentielle primaire.
4. Leucémie éosinophile chronique.
5. Leucémie neutrophile chronique.
6. Mastocytose.
7. Néoplasmes myéloprolifératifs inclassables.

Les syndromes myélodysplasiques désignent des troubles caractérisés par une production cellulaire inefficace et une dysplasie, avec un risque variable de transformation en leucémie aiguë. La cellularité de la moelle est souvent augmentée mais très variable. Il y a maturation mais aussi dysplasie d’une ou plusieurs lignées myéloïdes. L’hématopoïèse n’est pas efficace et il existe donc des cytopénies. Cet élément comprend :
1. Cytopénie réfractaire avec dysplasie en une ligne *.
• Anémie réfractaire.
• Neutropénie réfractaire.
• thrombocytopénie réfractaire.
2. Anémie réfractaire avec sidéroblastes annulaires.
3. Cytopénie réfractaire avec dysplasie de lignées multiples.
4. Anémie réfractaire avec excès de blastes.
5. Syndrome myélodysplasique avec d (5q).
6. Syndrome myélodyspastique inclassable.
7. Le syndrome myélodysplasique juvénile comprend une entité provisoire appelée cytopénie réfractaire juvénile.
Les syndromes myélodysplasiques / myéloprolifératifs comprennent des troubles dans lesquels coexistent des caractéristiques dysplasiques et prolifératives. Ce groupe comprend la leucémie juvénile myélomonocytaire, qui est représentative des deux syndromes (myélodysplasique et myéloproliférative). Près de la moitié des patients présentent un nombre de neutrophiles normal ou faible et une dysplasie de lignées cellulaires multiples sans organomégalie et moelle osseuse avec une morphologie ressemblant à une anémie réfractaire avec excès de blastes, mais avec monocytose. D’autres patients présentent une neutrophilie sévère, une monocytose et une splénomégalie. On ne sait pas encore s’il s’agit de 2 maladies différentes, une myélodysplasique et une myéloproliférative; cependant, jusqu’à présent, il n’y a pas de différences dans les anomalies cytogénétiques ou dans les schémas de croissance des colonies in vitro ou dans leur évolution clinique, il existe donc une controverse entre cliniciens et pathologistes en fonction de leur place dans la classification. Selon la dernière révision, dans cette catégorie se trouvent:
1. Leucémie myélomonocytaire chronique.
2. Leucémie myéloïde chronique atypique (BCR / ABL négative).
3. Leucémie myélomonocytaire juvénile.
4. Syndrome myélodysplasique / myéloprolifératif non classable.
Dans la catégorie des leucémies aiguës mieloblásticas (LAM) (qui est définie par un pourcentage supérieur à 20% de myéloblastes dans la moelle osseuse ou le diluant sanguin périphérique, ou la présence d’une anomalie cytogénétique en particulier, malgré le compte de blastes) reconnaît les groupes suivants:
1. LAM avec translocations cytogénétiques récurrentes.
2. LAM avec des caractéristiques myélodysplasiques.
3. LAM et MDS liés aux traitements antinéoplasiques.
4. LAM non classable.
5. Sarcome myéloïde.
6. Proliférations myéloïdes liées au syndrome de Down.
7. Néoplasme blastique plasmacitoïde des cellules dendritiques.
Les NÉOPLASMES LYMPHOÏDES
Sont ceux qui proviennent de cellules qui se développent normalement en lymphocytes T (LT cytotoxiques, collaborateurs ou régulateurs) ou en lymphocytes B (lymphocytes ou plasmocytes). En général, les néoplasmes lymphoïdes sont divisés en ceux dérivés des précurseurs lymphoïdes et ceux des lymphocytes matures et des plasmocytes et sont ensuite regroupés selon leur lignée (B ou T).
Historiquement, les néoplasmes lymphoïdes qui se produisent dans la moelle osseuse et impliquent la moelle osseuse ont été séparés de ceux qui se présentent sous forme de tumeur (lymphome). Cependant, il est maintenant connu que tout lymphome peut présenter des caractéristiques cliniques de leucémie et que toute leucémie peut occasionnellement se présenter sous forme de tumeur (sarcome granulocytaire). Dans la classification de l’OMS, le diagnostic de plusieurs néoplasmes lymphoïdes dépend non seulement de la localisation anatomique des cellules tumorales, mais également de l’origine morphologiquement définie de la cellule tumorale. Ces considérations ont annulé la pertinence des termes L1 et L2 de la classification FAB, car ils ne sont pas en corrélation avec leur immunophénotype, leurs anomalies génétiques ou leur évolution clinique (Figure 8). L3 est équivalent au lymphome de Burkitt en phase leucémique et doit être diagnostiqué comme tel.

1. Néoplasmes précurseurs. Il existe un consensus selon lequel les néoplasmes précurseurs qui se présentent sous forme de tumeurs solides et ceux impliquant la moelle osseuse et le sang sont biologiquement la même maladie avec des présentations cliniques différentes. La plupart des néoplasmes lymphoïdes précurseurs sont présentés comme des leucémies, il a donc été convenu que la classification devrait conserver le terme LAL pour la phase leucémique des néoplasmes précurseurs de type B et T. Il existe 2 catégories principales:
• Leucémies / lymphomes précurseurs B.
•Leucémies/lymphomes précurseurs T.
2. Néoplasmes à cellules B matures. La classification proposée considère les lymphomes et les leucémies du même type cellulaire comme la même maladie avec des présentations ou des stades cliniques différents. Les maladies spécifiques dérivées des cellules B matures sont les suivantes:
1. Leucémie lymphocytaire chronique / lymphome à petits lymphocytes.
2. Lymphome lymphoplasmocytaire.
3. Lymphome à cellules du manteau.
4. Leucémie prolymphocytaire à cellules B.
5. Lymphome folliculaire.
6. Lymphome diffus à grandes cellules B.
• Lymphome intravasculaire à grandes cellules B.
• Lymphome médiastinal primaire à grandes cellules B.
* Lymphome à grandes cellules B (lié au virus d’Epstein Barr – EBV).
••Lymphome à grandes cellules B riche en histiocytes et en lymphocytes T.
* Lymphome diffus à grandes cellules B du système nerveux central.
•*Lymphome cutané primaire diffus à grandes cellules B.
• Lymphome diffus à grandes cellules B des personnes âgées positif au VEB.
•Lymphome pasmablastique.
•Lymphome pleural primaire.
•* Lymphome à grandes cellules alkome positif (ALK).
•Lymphome de Burkitt.
7. Lymphome à cellules B de la zone marginale.
8. Lymphome à cellules B de la zone marginale extranodale.
9. Lymphome à cellules B de la zone marginale splénique
10. Leucémie à cellules velues.
11. Plasmocytome / myélome plasmocytaire.

NÉOPLASMES DES LIGNÉES MYÉLOÏDES ET LYMPHOÏDES
Certains néoplasmes expriment des marqueurs des lignées myéloïdes et lymphoïdes.indifférenciées) ou présentent des caractéristiques des deux lignées (phénotype mixte ou leucémie aiguë mixte).
Tableau 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍA
Bassan R, Hoelzer D. Thérapie moderne de la leucémie lymphoblastique aiguë. J Clin Oncol. 2011;29:523-43.
Burnett A, Wetzler M, Löwenberg B. Progrès thérapeutiques dans la leucémie aiguë. J Clin Oncol. 2011;29:487-94.
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Sang. 12 mai 2011; 117 (19): 5019-32. Epub 2011 7 février. Examen.
Cortes J, Hochhaus A, Hugues T, Kantajian H. Thérapies de première ligne et de sauvetage avec Inhibiteurs de la Tyrosine Kinase et autres traitements dans la leucémie myéloïde chronique. J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologie, Stratification des risques et Thérapie de la leucémie aiguë pédiatrique: Une mise à jour. J Clin Oncol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado MR. Évaluation indépendante multicentrique de notrevient chez les patients atteints de leucémie myéloïde chronique traités par Imatinib. Revue Institut National du Cancer. 2011;103:1-9.
Grever M, Lozanki G. Stratégies Modernes pour la leucémie à Cellules Velues. J Clin Oncol. 2011:29;583-90.
Gribben JG, O’Brien S. Mise à jour sur le traitement de la leucémie lymphoïde chronique. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Leucémie myéloïde chronique. Concepts actuels en Physiopathologie et traitement. Cancérologie. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib comparé à Imatinib/Cytarabine pour le traitement de première intention de la leucémie myéloïde chronique positive au chromosome Philadelphie précoce. Results of a Randomized Clinical trial of the Mexican Collaborative Leukemia Group. Leucémie clinique. 2008: 2(2);1128-32.
Lichtman MA. Classification et manifestations cliniques des troubles myéloïdes clonaux. En : Williams. Hématologie. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Génétique moléculaire de la leucémie myéloïde aiguë chez l’adulte. Implications pronostiques et thérapeutiques. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rév. 2010; 53:36-43.
Sanz M, Lo-Coco F. Approches modernes du traitement de la leucémie promyélocytaire aiguë. J Clin Oncol. 2011;29:495-503.
Remarque
* Dysplasie. Il fait référence à l’altération cytomorphologique qui comprend la dissociation de la maturation noyau-cytoplasme (rappelez-vous que la maturation de la chromatine dépend du cytoplasme de l’ADN et de l’ARN, donc le noyau s’arrête à la maturation pendant que le cytoplasme continue son processus normal) qui produit des cellules non viables et il y a apoptose intramédullaire.