Übersichtsartikel
Leukämie-für den Hausarzt
Leukämie für den Hausarzt
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
Leiter der Abteilung für Hämatologie. Krankenhaus Ángeles del Pedregal. Mexiko, DF. E-Mail-Adresse: [email protected]
b Innere Medizin. Krankenhaus Ángeles del Pedregal. Mexiko. DF.
c Innere Medizin. Krankenhaus Ángeles del Pedregal. Mexiko. DF.
Empfangen: 17.Oktober 2011
Akzeptiert: 07. Januar 2012
EINLEITUNG
Trotz der großen molekularen und therapeutischen Fortschritte bei der Untersuchung von Leukämien sind die grundlegenden Aspekte dieser Erkrankung dem Nicht-Hämatologen noch nicht klar bekannt.

DEFINITION
Leukämie ist der Begriff, der verwendet wird, um eine Gruppe von bösartigen Blutkrankheiten zu definieren. Eine frühzeitige Diagnose ist unerlässlich, da der Patient frühzeitig mit dem Hämatologiespezialisten in Kontakt treten kann, der den Diagnoseprozess leitet und die spezifische Behandlung anbietet. Es zeichnet sich durch eine klonale, autonome und abnormale Proliferation der Zellen aus, aus denen der Rest der normalen Blutzellen hervorgeht (Tumorverhalten im Allgemeinen). Dies impliziert, dass eine frühe Zelle eine genetische Veränderung erfährt, die dazu führt, dass ein abnormaler Klon (Kolonie) von sich selbst unkontrolliert auftritt. Diese abnormale Produktion ist gestört, da sich die abnormalen Zellen in Bild und Ähnlichkeit von sich selbst vermehren, so dass sie allmählich den Raum des normalen Knochenmarks einnehmen und eine fortschreitende Anämie, abnormale Blutungen und eine Prädisposition für Infektionen verursachen. Auf der anderen Seite, wenn abnormale Zellen in andere Gewebe eindringen, wird es zu einem Versagen der Funktion des betreffenden Organs kommen, zum Beispiel könnte sich eine Infiltration des Zentralnervensystems, die bei akuter lymphoblastischer Leukämie (LAL) auftritt, mit Kopfschmerzen, Krampfanfällen, fokussierten motorischen Veränderungen, erhöhtem Hirndruck und Versagen, eine frühzeitige Diagnose zu stellen und eine angemessene Behandlung bereitzustellen, zu Funktionsverlust und irreversiblen Folgen führen.
KLINISCHE MANIFESTATIONEN
Das klinische Bild ist vielfältig und hängt von der Art der Leukämie ab: akut oder chronisch, jedoch für die 2 gibt es unspezifische klinische Manifestationen (die bei jeder Krankheit auftreten):
1. Müdigkeit.
2. Leichte Müdigkeit.
3. Generalisierte Schwäche.
4. Wünsche ruhen oder im Bett zu bleiben.
5. Es erfordert die Hilfe von jemandem, um Ihre persönlichen Bedürfnisse zu erfüllen.
Chronische Leukämien sind träge und bis zu 50% der Fälle werden in einer routinemäßigen klinischen oder Laboruntersuchung bei Freiwilligen entdeckt, die als gesund gelten und Blut spenden.

Bei akuten Formen ergeben sich spezifische Manifestationen aus einem Mangel einer der Zelllinien:
1. Erythrozyten: anämisches Syndrom, dessen Intensität unabhängig vom Grad der Anämie vom Grad der Hypoxämie abhängt. Dyspnoe mittlerer Anstrengung bis zur Orthopathie.
2. Thrombozyten: Petechien, Ekchymose in den Extremitäten und in schwereren generalisierten Fällen trockene und nasse Blutung mit Epistaxis, Gingivorrhagie, Hämaturie, Mähne oder Hämatochesie. Sehr schwer im zentralen Nervensystem (ZNS).
3. Leukozyten: Fieber, Diaphorese, lokalisierte Infektionen bis hin zu offener Septikämie (Bakterien oder Pilze). Sie treten bei einer Neutropenie von weniger als 250 Gesamtneutrophilen / mm3 auf.
Infiltratives Syndrom: bezieht sich auf abnormale Implantation in jedem Gewebe, obwohl es häufig ist:
1. Hepatomegalie oder Splenomegalie (Abbildung 5).
2. Adenomegalie (lokal oder generalisiert).
3. Leukämischer Teint.
4. Knochenschmerzen durch Knochenmarksexpansion.
5. Weichteile (granulozytäres Sarkom).
6. Hoden.
7. SNC.
8. Zahnfleisch und jede Website (Abbildung 1).

Stoffwechselstörungen: sie resultieren aus einer abnormalen Hyperproduktion maligner Zellen und einer erhöhten Apoptose.
1. Lactatazidose.
2. Erhöhte Laktatdehydrogenase (LLD).
3. Hyperkaliämie.
4. Hyperurikämie.
5. Erhöhtes β2-Mikroglobulin.

Die klinische Evidenz überwiegt als Eckpfeiler der Verdachtsdiagnosen akuter Leukämien und jeglicher Erkrankungen, aber es folgt die Ergänzung der Diagnose mit Unterstützung des klinischen Labors bei der Zytometrie komplettes Blutbildoder speziell, dh die gründliche Beobachtung des peripheren Blutausstrichs durch das technische Personal, das die Vorbereitung des Abstrichs hat bei der Identifizierung von abnormalen Zellen, insbesondere Leukämie.

Laboränderungen, die einer besonderen Überprüfung bedürfen, umfassen:
1. Anämie (jeder Grad).
2. Leukopenie oder Leukozytose (Vorherrschen einer Zelllinie).
3. Thrombozytopenie.
4. Kombinationen: Bicitopenie oder Panzytopenie.
Besondere Vorsicht ist geboten, wenn das Labor das Vorhandensein von Leukozyten oder atypischen Lymphozyten (möglicherweise leukämische Blasten) meldet. Es ist ratsam, eine Expertenbewertung anzufordern (Abbildung 2).

Die Knochenmarkaspiration ist für die Diagnose unerlässlich (Abbildung 3), und 20% der Blasten sind erforderlich, um die Kriterien für akute Leukämie in einer ihrer Varianten festzulegen.

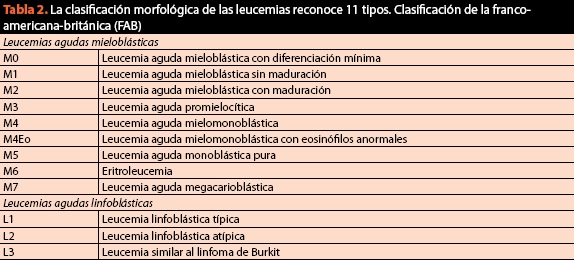
Im selben Verfahren sollten Proben für die endgültige Klassifizierung des Zustands und des Karyotyps und Immunphänotyps erhalten werden, da derzeit das zytomorphologische Kriterium von entscheidender Bedeutung ist, aber nicht mehr ausreicht. Die aktuelle und aktuelle Klassifikation maligner Blutkrankheiten ist in Tabelle 2 aufgeführt.
Die Behandlung zielt auf 2 wichtige Aspekte ab: Der erste ist der spezifische antileukämische und basiert auf der Verwendung von Arzneimitteln chemischen Ursprungs, die als Chemotherapie bekannt sind, deren Hauptziel es ist, alle Leukämiezellen aus dem Körper auszurotten, dh zu eliminieren. Der zweite Aspekt der Behandlung ist die Unterstützung von Komplikationen, die normalerweise bei Patienten bei der Aufnahme auftreten.
Die Behandlung zielt auf 2 wichtige Aspekte ab: Der erste ist der spezifische antileukämische und basiert auf der Verwendung von Arzneimitteln chemischen Ursprungs, die als Chemotherapie bekannt sind und deren Hauptziel es ist, alle Leukämiezellen aus dem Körper zu beseitigen.
Der zweite Aspekt der Behandlung ist die Unterstützung der Komplikationen, die Patienten normalerweise bei der Aufnahme aufweisen, wie zum Beispiel:
1. Anämie.
2. Abnormale Blutungen.
3. Pulmonale und generalisierte Infektionen, unter anderem (Abbildung 4).
4. Alle anderen angrenzenden Komplikationen, die der Patient haben kann (Komorbidität), wie Vorerkrankungen, z. B. Diabetes, Bluthochdruck, Herzerkrankungen und andere Krankheiten, die bei Patienten mit Leukämie häufig auftreten.


Daher ist es sehr wichtig zu berücksichtigen, dass die Behandlung gegen Leukämie multidisziplinär ist und andere Spezialisten wie die Unterstützung des Hämatologen beteiligt sind.
Die antileukämische Behandlung wird auch für verschiedene Arten von Leukämie und für akute Formen unterschiedlich sein. Es ist in 3 Phasen unterteilt:
1. Induktion der Remission. Ziel ist es, eine vollständige Remission (CR) zu erreichen, dh eine Normalisierung der Blutwerte des Patienten, das Fehlen von Symptomen oder Anzeichen dafür, dass Leukämie bei Infiltration anhält. Während des Prozesses sollte der Patient einen „leukämiefreien Zustand“ im Knochenmark haben und die Zukunft sollte die Erholung zu einer normalen Hämatopoese sein, und leider erholen sie sich in anderen Fällen mit der Krankheit, die von resistenter Leukämie spricht, deren Prognose schrecklich ist. Dieser erste Prozess kann 6 bis 8 Wochen dauern, um CR zu erreichen.
2. Konsolidierung. Es handelt sich um die Verwendung derselben Arzneimittel, die bei der Induktion oder Kombination anderer Chemotherapeutika verwendet wurden, auch um die Ausrottung verbleibender maligner Zellen zu verfolgen, die eine Resistenz gegen Erstanwendungen entwickeln könnten.
3. Wartung. Es ist bevorzugt, den Patienten unter der Wirkung einer Chemotherapie zu halten, bevor die Möglichkeit einer beginnenden Leukämieaktivität besteht, und dass er bei der Behandlung die Wirkung beibehält, bis die Krankheit verschwindet.

Bisher sind bei akuten Formen die strengsten Kriterien, die zu berücksichtigen sind, komplexer geworden, da es am besten ist, nach einer molekularen Remission zu suchen, bei der nach der anfänglichen chromosomalen Veränderung gesucht wird, wie sie bei promyelozytischer Leukämie (M3-FAB) mit t (15;17) zunächst sollte trotz CR nach spezifischen molekularen und zytogenetischen Studien gesucht werden, da bei anhaltender Translokation die aggressive Behandlung bis zur vollständigen Elimination des Klons fortgesetzt werden sollte. Diese Art von Leukämie sollte als potenziell heilbar angesehen werden und ist eine der häufigsten in der Latino-Bevölkerung.
Die Heilung der Krankheit hängt dann von der Eliminierung aller vorhandenen malignen Zellen im Patienten ab. Im Allgemeinen können einige Leukämien mit einer Chemotherapie allein geheilt werden, aber heute müssen sogenannte Prognosefaktoren, die auf mathematischen Modellen basieren, die es ermöglichen, Patienten in den Grad ihrer Prognose zu versetzen, große Bedeutung beigemessen werden und umfassen:
1. Die Art der Leukämie.
2. Die anfängliche molekulare Veränderung und ihre Persistenz trotz Behandlung oder Eradikation.
3. Alter. Patienten, die älter als 60 Jahre sind, haben im Vergleich zu jüngeren Patienten eine schlechte Prognose.
4. Chemotherapie. Die angegebenen Arzneimittel und vor allem die empfohlenen Dosen sollten verwendet werden. Zum Beispiel erreicht bei erwachsenen LAL die Verwendung des HyperCyVAD-Schemas (eskalierte Dosen von Cyclophosphamid, Vincristin, Adriamycin, Dexamethason in Kombination mit Cytosinarabinosid und Methotrexat) 90% CR und heilt in 50% der Fälle, Daten, die zuvor nicht mit anderen Schemata gesehen wurden. Diese Behandlung ist toxisch und erfordert, dass sie nur in Einrichtungen angewendet wird, die über ausreichende Unterstützungsressourcen verfügen. Leider haben in unserer Umgebung nicht alle Zentren die empfohlenen Medikamente und die Ergebnisse werden nicht reproduzierbar sein, da sie den Patienten während der Phase der maximalen Myelosuppression nicht ausreichend unterstützen.
5. Unterstützende Therapie. Die Errungenschaften der Chemotherapie und neue Medikamente, neue Kombinationen und mit mehr Spezifität zwingen die Umsetzung der multidisziplinären Teams mit der Adresse des Hämatologen; Installation und Verwendung von Zentralvenenkathetern; Unterstützung für Blutbank zur Unterstützung von Transfusionen von Blutplättchen und Erythrozyten (auch Produkte sternförmig); die Intervention des Infektiologen zum Nachweis von Infektionen und die angemessene Verwendung von Antibiotika oder Antimykotika, entweder prophylaktisch oder therapeutisch, falls erforderlich; die Bereitstellung von Isolationsräumen mit Wartungs- und Quartiermeisterdiensten, die eine bakterienfreie Umgebung einschließlich steriler Fütterung ermöglichen; ein Labor mit Patientenprobenmanagementprotokoll in spezifischen Behandlungsprotokollen und die Verwaltung spezieller Proben (Herstellung von Leukozytenkonzentraten in der Buffy-Coat-Hämatikzytometrie und Erzielung einer optimalen Ablesung); bereich der Herstellung von hochspezialisierten Medikamenten, und auf der anderen Seite ist die wichtigste die Pflege, Hilfs- und Verwaltungspersonal, die mit der ganzen Gruppe die Ergebnisse, die sie in anderen Ländern haben erwartet.
6. Knochenmarktransplantation (MO). Es ist eine komplexe und kostengünstige Art der Behandlung, die einen kompatiblen Blutspender und einen inaktiven Zustand mit einer hohen Wahrscheinlichkeit eines frühen oder späten Rückfalls oder mit Faktoren schlechter Prognose erfordert. Es ist ein Verfahren mit kurativeren Tendenzen, da es Megadosen der Chemotherapie verwendet, um die Leukämiezellen auszurotten, aber in dem Versuch, es auch die normalen Vorläufer auszurotten und der Ersatz eines neuen kompatiblen normalen Marks notwendig ist. Der Typ allogene (Geschwister kompatibel identisch) ist die beste Wahl, da der Empfänger akzeptiert nur durch Identität teilweise (die nur der totalen Akzeptanz sind eineiige Zwillinge), und daher führt zu einer Ablehnung der Graft-versus-host-Krankheit, was wiederum führt zu Graft-versus-Leukämie und erhöht die Ausrottung der Klone leukämischen, jedoch in schweren Fällen des Syndroms (Grad 3-4) Morbidität und Mortalität ist trotz der spezifischen Handhabung erhöht.
CHRONISCHE LEUKÄMIEN
1. Chronische lymphatische Leukämie. Es tritt häufiger bei älteren Menschen auf und das Kriterium ist die Persistenz der Lymphozytose von mehr als 10 x 109 / l und MO mit Infiltration von mehr als 50% der Lymphozyten mit CD5 + -Phänotyp. Das Kriterium der Behandlung ist die Verdoppelung des Kontos von Lymphozyten in einem Jahr oder Progression von Adenomegalie oder Splenomegalie, obwohl einige Fälle über diesen Standard hinaus durch das Vorhandensein von hämolytischer Anämie oder Thrombozytopenie, Autoimmun, und dann die Behandlung basierend auf der Kombination von Fludarabin, Cyclophosphamid und Prednison, in den Stadien I und II nur somenter und beobachtete und erwartete Entwicklung ohne Behandlung.
2. Chronische myeloische Leukämie (CML). Bei dieser Krankheit gibt es einen großen Fortschritt in der Kenntnis des Vorhandenseins des Philadelphia-Chromosoms, das 1950 in der Stadt der Amerikanischen Union beschrieben wurde, und dass seine Entstehung das erste Marker-Chromosom in Verbindung mit Malignität war, jedoch im Laufe der Jahre der Forschung gelang es uns, das t(9;22) mit der funktionellen Expression des Chromosoms mit der Produktion eines Onkoproteins mit großer Thyrokinokinase-Aktivität, das die Zellproliferation erhöht und was wiederum die große Leukozytose und Thrombozytose erklärt, mit der diese Patienten auftreten, sowie die große Splenomegalie (Abbildung 5).
Zu diesem Zeitpunkt sind 10 bis 12 Jahre seit der Entdeckung eines kleinen Moleküls vergangen, das speziell gegen dieses Phosphat-Donor-Molekülsubstrat für die interne Regulation der Leukämiezelle und ihrer Substrate gerichtet ist. Die ursprünglich als STI (Signal Transduction Inhibitor) erzeugte kompetitive Hemmung der Phosphorylierung, führte zur Zellapoptose und die Patienten erzielten klinische Ergebnisse, die noch nie zuvor mit molekularen Remissionen von bis zu 80-90% nach 10 Jahren gesehen wurden, was die natürliche Geschichte der Krankheit dramatisch veränderte, wobei die Vorbehandlung nicht länger als 3 Jahre dauerte. Diese Information verändert die Naturgeschichte und Prognose der Krankheit, die zuvor kurzfristig tödlich war.

Diese Beispiele sind eine Basis für die Fortschritte so intensiv, dass in den letzten Jahren aufgetreten und dass es wünschenswert ist, zu ermöglichen, provozieren das Interesse der Medizinstudenten, ihre Lehrer, vor allem aber der Bildungsbehörden der Leukämie im Allgemeinen, die die ersten 5 Plätze der Häufigkeit der malignen Erkrankung des Erwachsenen und der ersten Plätze bei den Kindern einnimmt, so dass die Hämatologie in den Kernfächern in den Lehrplan der Karriere der Medizin und sogar als Teil der der Kurse postgraduale Spezialisierung. Obwohl die britisch-französischamerikanische Klassifikation (FAB) für ihre Einfachheit nützlich ist, kann sie in bis zu 20% der Fälle zu diagnostischen und damit therapeutischen Fehlern führen. Aus diesem Grund ist die Klassifizierung nach immunhistochemischen und molekularbiologischen Methoden zu einer unabdingbaren Voraussetzung für die korrekte Klassifizierung und anschließende Behandlung von Patienten geworden (Abbildung 6).

Im Gegensatz zur Klassifikation FAB spiegelt die WHO (Tabelle 2) einen Paradigmenwechsel wider, durch den wir verstehen Krankheiten Blut, weil zum ersten Mal die genetische Information, die morphologische, zytochemische und immunphänotypische mit den klinischen Befunden innerhalb der diagnostischen Algorithmen von Neoplasmen des hämatopoetischen Gewebes kombiniert; die relative Bedeutung jedes Kriteriums unterscheidet sich zwischen Neoplasmen und es gibt keinen „Goldstandard“ für die Klassifizierung aller hämatologischen Malignome. Ziel war es, Entitäten zu definieren, die von Pathologen erkannt werden konnten und klinisch relevant waren. Seit seinem Erscheinen im Jahr 2001 wurden verschiedene Überarbeitungen vorgenommen, um den Inhalt in Bezug auf die aktuellsten Entdeckungen zu aktualisieren. Die letzte Überprüfung stammt aus dem Jahr 2008 und klassifiziert hämatologische Malignome wie folgt:
1. Myeloische Neoplasmen.
2. Lymphoide Neoplasmen.
3. Mastzell- oder Fettzellenkrankheiten.
4. Histiozytäre und dendritische Zellkrankheiten.

NEOPLASIE MYELOID
Myeloische Neoplasmen stammen von Vorläufern im Knochenmark ab und differenzieren sich in Erythrozyten, Granulozyten (Neutrophile, Basophile und Eosinophile), Monozyten und Megakaryozyten. Die FAB-Klassifizierung erkennt 3 Hauptkategorien an:
1. Akute myeloische Leukämie.
2. Myelodysplastische Syndrome.
3. Myeloproliferative Neoplasmen.
Die wichtigsten Determinanten der erkannten Kategorien sind morphologisch, histochemisch und immunphänotypisch und der Anteil der Blastenzellen, die Zelllinie und der Differenzierungsgrad der neoplastischen Zellen (Abbildung 7). In den letzten Jahren zeigten die genetischen Merkmale (zytogenetisch und molekular) sowie die Vorbehandlung und die Entwicklung der Myelodysplasie einen signifikanten Einfluss auf das klinische Verhalten dieser Leiden, die nicht immer gut mit den Kategorien der Krankheit korrelieren, so dass die zentrale Debatte für die Neuklassifizierung darin bestand, zwischen den Entitäten und pathologischen prognostischen Faktoren zu unterscheiden, um eine Klassifizierung mit klinischer Relevanz und Bedeutung für den Pathologen zu erreichen. Einige genetische Anomalien scheinen verschiedene Krankheiten zu definieren, während andere prognostische Faktoren für eine bestimmte Krankheit darstellen.

Derzeit gruppiert die WHO-Klassifikation myeloide Zustände in 4 Hauptgruppen:
1. Myeloproliferative Erkrankungen
2. Myelodysplastische Syndrome
3. Myelodysplastische/myeloproliferative Erkrankungen
4. Akute myeloische Leukämien

Myeloproliferative Erkrankungen sind eine Gruppe von klonalen Erkrankungen, die mit der Proliferation einer oder mehrerer myeloischer Linien verbunden sind. Es wird immer klarer, dass diese Krankheiten häufig mit Mutationen assoziiert sind, die zu abnormalen Anstiegen der Tyrosinkinaseaktivität und einer Wachstumsfaktor-unabhängigen Progenitorzellproliferation im Knochenmark führen. Der Prozentsatz der Blasten im Knochenmark ist normal oder leicht hoch, aber immer weniger als 20%. Die Hämatopoese ist normalerweise wirksam, was zu einer Erhöhung der Anzahl einer oder mehrerer reifer Zellen im peripheren Blut führt. Der Prototyp von myeloproliferativen Neoplasmen ist Philadelphia-Chromosom-positive (Ph1) chronische myeloische Leukämie (BCR / ABL). Die anderen Entitäten sind:
1. Polyzythämie vera.
2. Idiopathische Myelofibrose.
3. Primäre essentielle Thrombozythämie.
4. Chronische eosinophile Leukämie.
5. Chronische neutrophile Leukämie.
6. Mastozytose.
7. Nicht klassifizierbare myeloproliferative Neoplasmen.

Myelodysplastische Syndrome beziehen sich auf Störungen, die durch ineffektive Zellproduktion und Dysplasie mit einem variablen Risiko der Umwandlung in akute Leukämie gekennzeichnet sind. Die Zellularität im Knochenmark ist oft erhöht, aber sehr variabel. Es gibt Reifung, aber auch Dysplasie einer oder mehrerer myeloischer Linien. Hämatopoese ist nicht wirksam und daher existieren Zytopenien. Dieser Artikel enthält:
1. Refraktäre Zytopenie mit einzeiliger Dysplasie*.
• Refraktäre Anämie.
• Refraktäre Neutropenie.
* Refraktäre Thrombozytopenie.
2. Refraktäre Anämie mit ringförmigen Sideroblasten.
3. Refraktäre Zytopenie mit Dysplasie multipler Linien.
4. Refraktäre Anämie mit überschüssigen Blasten.
5. Myelodysplastisches Syndrom mit d (5q).
6. Nicht klassifizierbares myelodyspastisches Syndrom.
7. Das juvenile myelodysplastische Syndrom umfasst eine vorläufige Einheit, die als juvenile refraktäre Zytopenie bekannt ist.
Myelodysplastische / myeloproliferative Syndrome umfassen Erkrankungen, bei denen dysplastische und proliferative Merkmale nebeneinander bestehen. Diese Gruppe umfasst myelomonozytäre juvenile Leukämie, die für beide Syndrome (myelodysplastisch und myeloproliferativ) repräsentativ ist. Fast die Hälfte der Patienten weist normale oder niedrige Neutrophilenzahlen und Dysplasien multipler Zelllinien ohne Organomegalie und Knochenmark mit einer Morphologie auf, die einer refraktären Anämie mit überschüssigen Blasten ähnelt, jedoch mit Monozytose. Andere Patienten haben schwere Neutrophilie, Monozytose und Splenomegalie. Es ist noch nicht bekannt, ob es sich um 2 verschiedene Krankheiten handelt, eine myelodysplastische und eine myeloproliferative; bisher gibt es jedoch keine Unterschiede in den zytogenetischen Anomalien oder in den Wachstumsmustern der In-vitro-Kolonien oder in ihrer klinischen Entwicklung, so dass es Kontroversen zwischen Klinikern und Pathologen nach ihrem Platz innerhalb der Klassifikation gibt. Gemäß der letzten Revision befinden sich in dieser Kategorie:
1. Chronische myelomonozytäre Leukämie.
2. Atypische chronische myeloische Leukämie (BCR / ABL-negativ).
3. Juvenile myelomonozytäre Leukämie.
4. Myelodysplastisches / myeloproliferatives Syndrom nicht klassifizierbar.
In der Kategorie der akuten Leukämien mieloblásticas (LAM) (die durch einen Prozentsatz von mehr als 20% Myeloblasten im Knochenmark oder Blutverdünner peripheren definiert ist, oder das Vorhandensein einer zytogenetischen Anomalie insbesondere trotz der Berücksichtigung von Blasten) erkennt die folgenden Gruppen:
1. LAM mit rezidivierenden zytogenetischen Translokationen.
2. LAM mit myelodysplastischen Eigenschaften.
3. LAM und MDS im Zusammenhang mit antineoplastischen Behandlungen.
4. LAM nicht klassifizierbar.
5. Myeloisches Sarkom.
6. Myeloische Proliferationen im Zusammenhang mit dem Down-Syndrom.
7. Plasmacitoid blastisches Neoplasma von dendritischen Zellen.
LYMPHOIDE NEOPLASMEN
Sind solche, die aus Zellen stammen, die sich normalerweise zu T-Lymphozyten (zytotoxische ZELLEN, Kollaborateure oder Regulatoren) oder B-Lymphozyten (Lymphozyten oder Plasmazellen) entwickeln. Im Allgemeinen werden lymphoide Neoplasmen in solche unterteilt, die von lymphoiden Vorläufern und solchen aus reifen Lymphozyten und Plasmazellen stammen, und anschließend nach ihrer Abstammung (B oder T) gruppiert.
In der Vergangenheit wurden lymphoide Neoplasmen, die im Knochenmark auftreten und das Knochenmark betreffen, von denen getrennt, die sich als Tumor (Lymphom) präsentieren. Es ist jedoch bekannt, dass jedes Lymphom klinische Merkmale einer Leukämie aufweisen kann und dass jede Leukämie gelegentlich als Tumor (granulozytäres Sarkom) auftreten kann. In der WHO-Klassifikation hängt die Diagnose mehrerer lymphoider Neoplasmen nicht nur von der anatomischen Lage der Tumorzellen ab, sondern auch von der morphologisch definierten Herkunft der Tumorzelle. Diese Überlegungen machten die Relevanz der Begriffe L1 und L2 der FAB-Klassifikation zunichte, da sie nicht mit ihrem Immunphänotyp, genetischen Anomalien oder ihrem klinischen Verlauf korrelieren (Abbildung 8). L3 entspricht dem Burkitt-Lymphom in der leukämischen Phase und sollte als solches diagnostiziert werden.

1. Vorläuferneoplasmen. Es besteht ein Konsens darüber, dass Vorläuferneoplasmen, die als solide Tumoren auftreten, und solche, die Knochenmark und Blut betreffen, biologisch dieselbe Krankheit mit unterschiedlichen klinischen Darstellungen sind. Daher wurde vereinbart, dass die Klassifizierung den Begriff LAL für die leukämische Phase von Vorläuferneoplasmen vom Typ B und T beibehalten sollte. Es gibt 2 Hauptkategorien:
• Vorläufer B Leukämien / Lymphome.
• Vorläuferleukämien/Lymphome T.
2. Reife B-Zell-Neoplasmen. Die vorgeschlagene Klassifikation betrachtet Lymphome und Leukämien desselben Zelltyps als dieselbe Krankheit mit unterschiedlichen klinischen Darstellungen oder Stadien. Spezifische Krankheiten, die von reifen B-Zellen abgeleitet sind, sind wie folgt:
1. Chronische lymphatische Leukämie / kleines Lymphozytenlymphom.
2. Lymphoplasmazytisches Lymphom.
3. Mantelzell-Lymphom.
4. Prolymphozytäre B-Zell-Leukämie.
5. Follikuläres Lymphom.
6. Diffuses großzelliges Lymphom B.
• Intravaskuläres großzelliges Lymphom B.
• Primäres mediastinales großzelliges Lymphom B.
• Großzelliges Lymphom B (verwandt mit dem Epstein-Barr-Virus-EBV).
•* Histiozyten- und T-Zell-reiches großes B-Zell-Lymphom.
* Diffuses großes B-Zell-Lymphom des Zentralnervensystems.
• * Diffuses primäres kutanes großzelliges B-Zell-Lymphom.
• Diffuses großzelliges B-Zell-Lymphom älterer Menschen positiv für EBV.
• Pasmablastisches Lymphom.
• Primäres Pleura-Lymphom.
• * Alkoma-positives (ALK) großzelliges Lymphom.
• Burkitt-Lymphom.
7. Randzone B-Zell-Lymphom.
8. Extranodale Randzone B-Zell-Lymphom.
9. Milz-Randzone B-Zell-Lymphom
10. Haarzell-Leukämie.
11. Plasmozytom / Plasmazell-Myelom.

MYELOISCHE UND LYMPHOIDE LINIENNEOPLASMEN
Einige Neoplasmen exprimieren Marker sowohl der myeloischen als auch der lymphoiden Linien.b. undifferenziert) oder vorhandene Merkmale beider Linien (Mischphänotyp oder Mischlinie akute Leukämie).
Tabelle 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍA
Bassan R, Hoelzer D. Moderne Therapie der akuten lymphoblastischen Leukämie. In: J Clin Oncol. 2011;29:523-43.
Burnett A, Wetzler M, Löwenberg B. Therapeutische Fortschritte bei akuter Leukämie. In: J Clin Oncol. 2011;29:487-94.
Stein H, Pileri S,Campo E, Swerdlow SH, Harris NL, Jaffe ES. Die 2008 WHO-Klassifikation von lymphatischen Neoplasmen und darüber hinaus: sich entwickelnde Konzepte und praktische Anwendungen. Blut. 2011 Mai 12;117(19): 5019-32. Epub 2011 Februar 7. Bewertung. Cortes J, Hochhaus A, Hugues T, Kantajian H. Front-Line- und Salvage-Therapien mit Tyrosinkinase-Inhibitoren und anderen Behandlungen bei chronischer myeloischer Leukämie. In: J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologie, Risikostratifizierung und Therapie der pädiatrischen akuten Leukämie: Ein Update. In: J Clin Oncol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado Mr. Multizentrische unabhängige Bewertung unserer Ergebnisse bei Patienten mit chronischer myeloischer Leukämie, die mit Imatinib behandelt wurden. Zeitschrift Nationales Krebsinstitut. 2011;103:1-9.
Grever M, Lozanki G. Moderne Strategien für Haarzell-Leukämie. In: J Clin Oncol. 2011:29;583-90. Gribben JG, O’Brien S. Update zur Therapie der chronischen lymphatischen Leukämie. In: J Clin Oncol. 2011;29:544-50.
Hurtado HERR, Vargas VP, Cortes FJ. Chronische myeloische Leukämie. Aktuelle Konzepte in Physiopathologie und Behandlung. Cancerología. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib im Vergleich zu Imatinib/Cytarabin zur Erstlinientherapie der frühen Philadelphia-Chromosom-positiven chronischen Myeloischen Leukämie. Ergebnisse einer randomisierten klinischen Studie der mexikanischen kollaborativen Leukämie-Gruppe. Klinische Leukämie. 2008: 2(2);1128-32. In:Lichtman MA. Klassifikation und klinische Manifestationen der klonalen myeloischen Erkrankungen. In: Williams. Hämatologie. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Molekulare Genetik der erwachsenen akuten myeloischen Leukämie. Prognostische und therapeutische Implikationen. In: J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rev Fac Med UNAM. 2010; 53:36-43.
Sanz M, Lo-Coco F. Moderne Ansätze zur Behandlung von akuter Promyelozytenleukämie. In: J Clin Oncol. 2011;29:495-503.
Hinweis
* Dysplasie. Es bezieht sich auf die zytomorphologische Veränderung, die die Dissoziation der Zellkern-Zytoplasma-Reifung einschließt (denken Sie daran, dass die Chromatinreifung vom DNA- und RNA-Zytoplasma abhängt, daher stoppt der Zellkern bei der Reifung, während das Zytoplasma seinen normalen Prozess fortsetzt), die nicht lebensfähige Zellen produziert und es gibt intramedulläre Apoptose.