granskningsartikel
leukemi-för allmänläkaren
leukemi för allmänläkaren
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
chef för avdelningen för hematologi. Sjukhus Asingeles del Pedregal. Mexiko, DF. E-postadress: [email protected]
b internmedicin. Sjukhus Asingeles del Pedregal. Mexico. DF.
C internmedicin. Sjukhus Asingeles del Pedregal. Mexico. DF.
mottagen: 17 oktober 2011
accepterad: 07 januari 2012
introduktion
trots de stora molekylära och terapeutiska framstegen i studien av leukemier är de grundläggande aspekterna av detta tillstånd ännu inte klart kända av icke-hematologen, så målet med detta arbete är att ge grundläggande information till medicinska studenter och läkare i allmänhet, och det gör framför allt att få allmän kunskap om leukemier, deras tidiga diagnos och söka tidig referens med hematologen.

definition
leukemi är termen som används för att definiera en grupp maligna blodsjukdomar. Tidig diagnos är nödvändig, eftersom det gör det möjligt för patienten att gå tidigt med hematologispecialisten, som kommer att leda diagnosprocessen och erbjuda den specifika behandlingen. Det kännetecknas av att ha en klonal, autonom och onormal proliferation av cellerna som ger upphov till resten av blodets normala celler (tumörbeteende i allmänhet).
detta innebär att en tidig cell genomgår en genetisk förändring som kommer att orsaka att en onormal klon (koloni) av sig själv uppträder okontrollerbart. Denna onormala produktion är störd eftersom de onormala cellerna multiplicerar i bild och likhet med sig själva, så de upptar gradvis utrymmet i den normala benmärgen och orsakar progressiv anemi, onormal blödning och predisposition till infektioner. Å andra sidan, när onormala celler invaderar andra vävnader, kommer det att misslyckas med det berörda organets funktion, till exempel infiltration i centrala nervsystemet som uppstår vid akut lymfoblastisk leukemi (LAL) kan manifestera med huvudvärk, anfall, fokuserade motoriska förändringar, ökat intrakraniellt tryck och misslyckande med att göra en tidig diagnos och ge adekvat behandling, kommer att medföra förlust av funktion och irreversibla konsekvenser.
kliniska manifestationer
den kliniska bilden är olika och beror på typen av leukemi: akut eller kronisk, men för 2 finns det icke-specifika kliniska manifestationer (som förekommer i någon sjukdom):
1. Trötthet.
2. Lätt trötthet.
3. Allmän svaghet.
4. Önskar att stanna vila eller i sängen.
5. Det kräver hjälp av någon för att möta dina personliga behov.
kroniska leukemier är indolenta och upp till 50% av fallen upptäcks i en rutinmässig klinisk eller laboratoriegranskning hos frivilliga som anses vara friska och kommer att donera blod, men när sjukdomen fortskrider presenteras ospecifika manifestationer men nu är de specifika (Tabell 1).

i akuta former härrör specifika manifestationer från brist på en av cellinjerna:
1. Erytrocyter: anemiskt syndrom vars intensitet beror på graden av hypoxemi oavsett graden av anemi. Andnöd av medium ansträngning tills orthoprea.
2. Blodplättar: petechiae, ekchymos i extremiteter och i mer allvarliga generaliserade fall, torr och våt blödning med epistaxis, gingivorragi, hematuri, man eller hematochesia. Mycket svår i centrala nervsystemet (CNS).
3. Leukocyter: feber, diaphoresis, lokaliserade infektioner upp till frank septikemi (bakterier eller svampar). De förekommer med neutropeni mindre än 250 Totala neutrofiler/mm3.
infiltrativt syndrom: avser onormal implantation i vilken vävnad som helst, även om det är vanligt:
1. Hepatomegali eller splenomegali (Figur 5).
2. Adenomegali (lokal eller generaliserad).
3. Leukemisk hudfärg.
4. Bensmärta från benmärgsutvidgning.
5. Mjuka vävnader (granulocytisk sarkom).
6. Testikel.
7. SNC.
8. Tandkött och vilken plats som helst (figur 1).

metaboliska störningar: de är resultatet av onormal hyperproduktion av maligna celler och ökad apoptos.
1. Acidos.
2. Ökat mjölkdehydrogenas (LLD).
3. Hyperkalemi.
4. Hyperurikemi.
5. Ökad β2-mikroglobulin.

de kliniska bevisen dominerar som hörnstenen i de misstänkta diagnoserna av akuta leukemier och av något tillstånd, men det som följer är att komplettera diagnosen med stöd av det kliniska laboratoriet i cytometry komplett blodantal, eller speciellt, det vill säga den grundliga observationen av perifert blodutstryk av den tekniska personalen som har förberedelse vid identifiering av onormala celler, särskilt leukemi.

laboratorieändringar som kräver särskild granskning inkluderar:
1. Anemi (någon grad).
2. Leukopeni eller leukocytos (övervägande av en cellinje).
3. Trombocytopeni.
4. Kombinationer: bicitopeni eller pancytopeni.
särskild försiktighet bör iakttas när laboratoriet rapporterar närvaron av leukocyter eller atypiska lymfocyter (kan vara leukemiska Blaster). Det är lämpligt att begära en expertrecension (Figur 2).

Benmärgsaspiration är avgörande för diagnos (Figur 3) och 20% av blasterna krävs för att fastställa kriterierna för akut leukemi i någon av dess sorter.

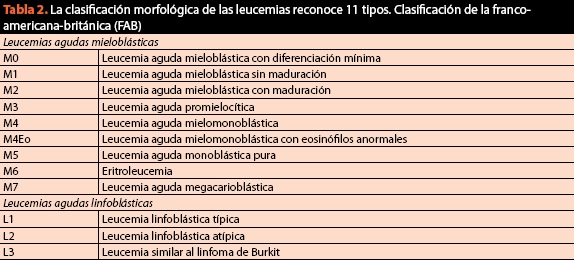
i samma förfarande bör prover erhållas för slutlig klassificering av tillståndet och begäran karyotyp och immunfenotyp, eftersom det cytomorfologiska kriteriet för närvarande är av vital betydelse men inte längre är tillräckligt. Den nuvarande och nuvarande klassificeringen av maligna blodsjukdomar anges i Tabell 2.
behandlingen riktar sig till 2 viktiga aspekter: den första är den specifika antileukemiska och baseras på användningen av läkemedel av kemiskt ursprung som kallas kemoterapi, vars huvudsyfte är att utrota, det vill säga eliminera alla leukemiska celler från kroppen. Den andra aspekten av behandlingen är stöd för komplikationer som vanligtvis uppstår hos patienter vid antagning.
behandlingen syftar till 2 viktiga aspekter: den första är den specifika antileukemiska och baseras på användningen av läkemedel av kemiskt ursprung som kallas kemoterapi, vars huvudsyfte är att utrota, det vill säga eliminera alla leukemiska celler från kroppen.
den andra aspekten av behandlingen är stödet för de komplikationer som patienter vanligtvis uppvisar vid antagning, såsom:
1. Järnbristanemi.
2. Onormal blödning.
3. Lung-och generaliserade infektioner, bland andra (Figur 4).
4. Eventuella andra intilliggande komplikationer som patienten kan ha (komorbiditet), såsom befintliga tillstånd, t.ex. diabetes, hypertoni, hjärtsjukdom och andra sjukdomar som är vanliga bland patienter som lider av leukemi.


därför är det mycket viktigt att ta hänsyn till att behandlingen mot leukemi är tvärvetenskaplig och involverar deltagande av andra specialister som stöd för hematologen.
Antileukemisk behandling kommer också att vara olika för olika typer av leukemi och för akuta former. Den är uppdelad i 3 faser:
1. Induktion av remission. Målet är att uppnå fullständig remission (CR), det vill säga normalisering av patientens blodvärden, frånvaron av några symtom eller tecken på att leukemi kvarstår med infiltration. Under processen ska patienten ha ETT” leukemifritt tillstånd ” i benmärgen och framtiden bör vara återhämtningen till en normal hematopoiesis, och tyvärr i andra fall återhämtar de sig med sjukdomen, som talar om resistent leukemi vars prognos är hemsk. Denna första process kan ta 6 till 8 veckor för att uppnå CR.
2. Konsolidering. Det handlar om användning av samma läkemedel som användes vid induktion eller kombination av andra kemoterapeutika, även för att följa utrotningen av kvarvarande maligna celler som kan utveckla resistens mot första användningen.
3. Underhåll. Det är att föredra att hålla patienten under effekten av kemoterapi före möjligheten till begynnande leukemisk aktivitet och att den med behandlingen upprätthåller effekten tills sjukdomen försvinner.

hittills för akuta former blir de strängaste kriterierna att överväga CR mer komplexa, eftersom det är bäst att leta efter molekylär remission, vilket innebär sökandet efter den initiala kromosomala förändringen som sker i fallet med promyelocytisk leukemi (M3-FAB) med t (15;17) initialt bör det sökas efter specifika molekylära och cytogenetiska studier trots att de är i CR, eftersom om translokationen kvarstår, bör den aggressiva behandlingen fortsättas tills klonens totala eliminering. Denna variation av leukemi bör betraktas som potentiellt härdbar och är en av de vanligaste i Latino-befolkningen.
botningen av sjukdomen beror då på eliminering av alla befintliga maligna celler i patienten. I allmänhet kan vissa leukemier vara mottagliga för botemedel med kemoterapi ensam, men idag måste mycket vikt ges till så kallade prognostiska faktorer som är baserade på matematiska modeller som gör att patienter kan placeras i graden av prognos de har och inkluderar:
1. Typ av leukemi.
2. Den initiala molekylära förändringen och dess uthållighet trots behandling eller utrotning.
3. ålder. Patienter äldre än 60 år har en dålig prognos jämfört med yngre patienter.
4. Kemoterapi. De angivna läkemedlen och framför allt de rekommenderade doserna ska användas. Till exempel, i vuxen LAL, uppnår användningen av HyperCyVAD-systemet (eskalerade doser cyklofosfamid, vinkristin, adriamycin, dexametason i kombination med cytosinarabinosid och metotrexat) 90% CR och botar i 50% av fallen, data som inte tidigare sett med andra system. Denna behandling är giftig och kräver att den endast används i institutioner som har tillräckliga stödresurser. Tyvärr har inte alla centra i vår miljö de rekommenderade medicinerna och resultaten kommer inte att reproduceras, eftersom de inte har tillräckligt med stöd för patienten under fasen med maximal myelosuppression.
5. Stödjande terapi. Resultaten av kemoterapi och nya läkemedel, nya kombinationer och med mer specificitet tvingar genomförandet av de tvärvetenskapliga lagen med hematologens adress; installation och användning av centrala venkatetrar; stöd för blodbank för stöd av transfusioner av blodplättar och erytrocyter (även produkter stellate); infektologens ingripande för detektering av infektioner och lämplig användning av antibiotika eller antifungala medel, antingen profylaktiska eller terapeutiska, om det behövs; tillhandahållande av isoleringsrum med underhålls-och quartermaster-tjänster som gör det möjligt att uppnå en bakteriefri miljö, inklusive steril utfodring; ett laboratorium med patientprovhanteringsprotokoll i specifika behandlingsprotokoll och hantering av speciella prover (beredning av leukocytkoncentrat i buffy coat hematisk cytometri och uppnå optimal avläsning); området för beredning av högspecialiserade Läkemedel, och å andra sidan är det viktigaste vård -, hjälp-och administrativ personal, som med hela gruppen förväntas de resultat de har i andra länder.
6. Benmärgstransplantation (MO). Det är en komplex och högkostnadstyp av behandling som kräver en kompatibel MO-givare och ett inaktivt tillstånd med stor sannolikhet för tidigt eller sent återfall eller med faktorer med dålig prognos. Det är ett förfarande med mer botande tendenser eftersom det använder megadoser av kemoterapi för att utrota leukemiska celler, men i försöket utrotar det också de normala föregångarna och ersättning av en ny kompatibel normal märg är nödvändig. Typen allogen (syskonkompatibel identisk) är det bästa urvalet eftersom mottagaren endast accepterar genom identitets partiell (den enda av total acceptans är identiska tvillingar) och resulterar därför i en avstötning av transplantat-mot-värd-sjukdomen, vilket i sin tur leder till transplantat-mot-leukemi och ökar utrotningen av klonerna leukemisk, men i svåra fall av syndromet (grad 3-4) ökar sjukligheten och dödligheten trots den specifika hanteringen.
kroniska leukemier
1. Kronisk lymfocytisk leukemi. Det förekommer oftare hos äldre människor och kriteriet är persistensen av lymfocytos på mer än 10 x 109/l och MO med infiltration av mer än 50% lymfocyter med CD5+fenotyp. Kriteriet för behandling är duplicering av kontot av lymfocyter i ett år eller progression av adenomegalia eller splenomegali, även om vissa fall utöver denna standard genom närvaron av hemolytisk anemi eller trombocytopeni, autoimmun, och indikerade sedan behandlingen baserad på kombinationen av fludarabin, cyklofosfamid och prednison, i steg i och II endast somange och observerad och förväntad utveckling utan behandling.
2. Kronisk myeloid leukemi (CML). I denna sjukdom finns det ett stort framsteg i kunskapen om närvaron av Philadelphia-kromosomen, som beskrevs 1950 i staden American Union, och att starten var den första markörkromosomen i samband med malignitet, men under årens forskning lyckades vi känna till t (9;22) Med det funktionella uttrycket av kromosomen med produktion av ett onkoprotein med stor tyrokinokinasaktivitet, vilket ökar cellproliferationen och som i sin tur förklarar den stora leukocytos och trombocytos som dessa patienter presenterar, liksom den stora splenomegali (Figur 5).
vid denna tidpunkt har 10 till 12 år gått sedan upptäckten av en liten molekyl specifikt riktad mot detta fosfat-donatormolekylära substrat för intern reglering av leukemisk cell och dess substrat. Den initialt kallade STI (signaltransduktionshämmaren) producerade konkurrenskraftig hämning av fosforyleringen, ledde till cellapoptos och patienterna uppnådde kliniska resultat som aldrig tidigare sett med molekylära remissioner på upp till 80-90% vid 10 år, vilket dramatiskt förändrade sjukdomens naturhistoria, med förbehandlingen var inte mer än 3 år. Denna information gör en förändring i sjukdomens naturhistoria och prognos, tidigare dödlig på kort sikt.

dessa exempel är en bas för framstegen så intensiva som inträffade under de senaste åren och att det är önskvärt att tillåta provocera intresse för medicinska studenter, deras lärare, men särskilt av utbildningsmyndigheterna i leukemi i allmänhet, som upptar de första 5 platserna för frekvensen av malign sjukdom hos vuxna och av de första platserna hos barnen, så hematologin ska ingå i kärnämnena i läroplanen för medicinens karriär och även som en del av kurserna forskarutbildning specialisering.
Även om den är användbar för sin enkelhet kan den brittiska fransk-amerikanska klassificeringen (FAB) leda till diagnostiska och därför terapeutiska fel i upp till 20% av fallen. Av denna anledning har klassificering med immunhistokemi och molekylärbiologiska metoder blivit ett nödvändigt krav för korrekt klassificering och efterföljande hantering av patienter (Figur 6).

till skillnad från klassificeringen FAB återspeglar WHO (tabell 2) en förändring i paradigmet genom vilket vi förstår sjukdomar blod, för första gången kombinerade den genetiska informationen, den morfologiska, cytokemiska och immunofenotypiska med de kliniska fynden inom de diagnostiska algoritmerna för neoplasmer i den hematopoietiska vävnaden; den relativa betydelsen av varje kriterium skiljer sig mellan neoplasmer och det finns ingen ”guldstandard” för klassificering av alla hematologiska maligniteter. Målet var att definiera enheter som kunde erkännas av patologer och som hade klinisk relevans. Sedan dess utseende 2001 har olika revideringar gjorts för att uppdatera innehållet i förhållande till de senaste upptäckterna. Den senaste översynen var från 2008 och klassificerar hematologiska maligniteter enligt följande:
1. Myeloida neoplasmer.
2. Lymfoida neoplasmer.
3. Mastcell eller fettcellsjukdomar.
4. Histiocytiska och dendritiska cellsjukdomar.

NEOPLASIS MYELOID
myeloida neoplasmer härrör från stamfäder i benmärgen, differentieras till erytrocyter, granulocyter (neutrofiler, basofiler och eosinofiler), monocyter och megakaryocyter. FAB-klassificeringen känner igen 3 huvudkategorier:
1. Akut myeloid leukemi.
2. Myelodysplastiska syndrom.
3. Myeloproliferativa neoplasmer.
de viktigaste determinanterna för de kategorier som erkänns med morfologisk, histokemisk och immunofenotypisk och andelen blastceller, celllinjen och graden av differentiering av neoplastiska celler (figur 7). Under de senaste åren visade de genetiska egenskaperna (cytogenetiska och molekylära), liksom förbehandlingen och utvecklingen av myelodysplasi, en signifikant inverkan på det kliniska beteendet hos dessa lidanden, som inte alltid korrelerar väl med FAB: s kategorier, så att den centrala debatten för omklassificering var att diskriminera mellan enheterna och patologiska prognostiska faktorer, för att uppnå en klassificering med klinisk relevans och betydelse för patologen. Vissa genetiska avvikelser verkar definiera olika sjukdomar, medan andra representerar prognostiska faktorer för en specifik sjukdom.

För närvarande grupperar WHO-klassificeringen myeloida tillstånd i 4 huvudgrupper:
1. Myeloproliferativa sjukdomar
2. Myelodysplastiska syndrom
3. Myelodysplastiska / myeloproliferativa sjukdomar
4. Akut myeloid leukemi

myeloproliferativa sjukdomar är en grupp klonala störningar associerade med proliferationen av en eller flera myeloida linjer. Det blir allt tydligare att dessa sjukdomar ofta är förknippade med mutationer som orsakar onormala ökningar av tyrosinkinasaktivitet och tillväxtfaktoroberoende progenitorcellsproliferation i benmärgen. Andelen blaster i benmärgen är normal eller något hög, men alltid mindre än 20%. Hematopoiesis är vanligtvis effektiv, vilket resulterar i en ökning av räkningen av en eller flera mogna celler i perifert blod. Prototypen av myeloproliferativa neoplasmer är Philadelphia kromosompositiv (Ph1) kronisk myeloid leukemi (BCR/ABL). De andra enheterna som ingår är:
1. Polycytemi vera.
2. Idiopatisk myelofibros.
3. Primär essentiell trombocytemi.
4. Kronisk eosinofil leukemi.
5. Kronisk neutrofil leukemi.
6. Mastocytos.
7. Oklassificerbara myeloproliferativa neoplasmer.

myelodysplastiska syndrom avser störningar som kännetecknas av ineffektiv cellproduktion och dysplasi, med varierande risk för omvandling till akut leukemi. Cellularitet i märgen ökar ofta men mycket varierande. Det finns mognad men också dysplasi av en eller flera myeloida linjer. Hematopoiesis är inte effektiv och därför finns cytopenier. Denna artikel innehåller:
1. Refraktär cytopeni med en linje dysplasi*.
• eldfast anemi.
• refraktär neutropeni.
• refraktär trombocytopeni.
2. Eldfast anemi med ringformiga sideroblaster.
3. Eldfast cytopeni med dysplasi av flera linjer.
4. Eldfast anemi med överskott av blaster.
5. Myelodysplastiskt syndrom med d (5q).
6. Oklassificerbart myelodyspastiskt syndrom.
7. Juvenil myelodysplastiskt syndrom, inkluderar en provisorisk enhet som kallas juvenil refraktär cytopeni.
myelodysplastiska / myeloproliferativa syndrom inkluderar störningar där dysplastiska och proliferativa egenskaper samexisterar. Denna grupp innefattar myelomonocytisk juvenil leukemi, som är representativ för båda syndromen (myelodysplastisk och myeloproliferativ). Nästan hälften av patienterna med normala eller låga neutrofilantal och dysplasi av flera cellinjer utan organomegali och benmärg med morfologi som liknar eldfast anemi med överskott av blaster, men med monocytos. Andra patienter har svår neutrofili, monocytos och splenomegali. Det är ännu inte känt om de är 2 olika sjukdomar, en myelodysplastisk och en myeloproliferativ; hittills finns det dock inga skillnader i cytogenetiska abnormiteter eller i tillväxtmönstren för in vitro-kolonierna eller i deras kliniska utveckling, så det finns kontroverser mellan kliniker och patologer beroende på deras plats inom klassificeringen. Enligt den senaste revisionen finns i denna kategori:
1. Kronisk myelomonocytisk leukemi.
2. Atypisk kronisk myeloid leukemi (BCR / ABL-negativ).
3. Juvenil myelomonocytisk leukemi.
4. Myelodysplastiskt / myeloproliferativt syndrom kan inte klassificeras.
i kategorin av akuta leukemier mielobly cygsticas (LAM) (som definieras med en procentandel större än 20% myeloblaster i benmärgen eller blodförtunnande perifera, eller närvaron av en cytogenetisk abnormitet i synnerhet, trots blasts konto) känner igen följande grupper:
1. LAM med återkommande cytogenetiska translokationer.
2. LAM med myelodysplastiska egenskaper.
3. LAM och MDS relaterade till antineoplastiska behandlingar.
4. LAM inte klassificerbar.
5. Myeloid sarkom.
6. Myeloid proliferation relaterad till Downs syndrom.
7. Plasmacitoid blastisk neoplasma av dendritiska celler.
lymfoida neoplasmer
är de som härrör från celler som normalt utvecklas till T-lymfocyter (cytotoxiska LT, kollaboratörer eller regulatorer) eller B-lymfocyter (lymfocyter eller plasmaceller). I allmänhet är lymfoida neoplasmer uppdelade i de som härrör från lymfoida prekursorer och de från mogna lymfocyter och plasmaceller och grupperas därefter enligt deras härstamning (B eller T).
historiskt sett har lymfoida neoplasmer som förekommer i benmärgen och involverar benmärgen separerats från de som förekommer som en tumör (lymfom). Det är emellertid nu känt att något lymfom kan uppvisa kliniska egenskaper hos leukemi och att någon leukemi ibland kan förekomma som en tumör (granulocytisk sarkom). I WHO-klassificeringen beror diagnosen av flera lymfoida neoplasmer inte bara på tumörcellernas anatomiska läge utan också på tumörcellens morfologiskt definierade ursprung. Dessa överväganden upphävde relevansen av termerna L1 och L2 i Fab-klassificeringen, eftersom de inte korrelerar med deras immunofenotyp, genetiska avvikelser eller med deras kliniska förlopp (figur 8). L3 motsvarar Burkitt lymfom i leukemisk fas och bör diagnostiseras som sådan.

1. Prekursor neoplasmer. Det finns enighet om att prekursorneoplasmer som presenterar som fasta tumörer och de som involverar benmärg och blod är biologiskt samma sjukdom med olika kliniska presentationer. De flesta prekursorlymfoida neoplasmer presenteras som leukemier, så det var överens om att klassificeringen skulle behålla termen LAL för leukemisk fas av typ B och t prekursor neoplasmer. Det finns 2 huvudkategorier:
• föregångare B leukemier / lymfom.
• föregångare leukemier / lymfom T.
2. Mogna B-cell neoplasmer. Den föreslagna klassificeringen beaktar lymfom och leukemier av samma celltyp som samma sjukdom med olika kliniska presentationer eller steg. Specifika sjukdomar som härrör från mogna B-celler är följande:
1. Kronisk lymfocytisk leukemi / litet lymfocytlymfom.
2. Lymfoplasmacytiskt lymfom.
3. Mantelcelllymfom.
4. Prolymphocytic B-cell leukemi.
5. Follikulärt lymfom.
6. Diffust storcellslymfom B.
• intravaskulärt storcellslymfom B.
• primärt mediastinalt storcellslymfom B.
• storcellslymfom B (relaterat till Epstein Barr-virus-EBV).
•• Histiocyt-och t-cellrikt stort B-celllymfom.
* diffus centrala nervsystemet stort B-celllymfom.
• * diffus primär kutan stor B-celllymfom.
• diffus stor B-celllymfom hos äldre positiva för EBV.
• Pasmablastiskt lymfom.
• primär pleural lymfom.
• * Alkomapositivt (ALK) storcellslymfom.
• Burkitt lymfom.
7. Marginalzon B-celllymfom.
8. Extranodal marginalzon B-celllymfom.
9. Mjältmarginalzon B-celllymfom
10. Hårig cell leukemi.
11. Plasmocytom / plasmacellmyelom.

myeloid och lymfoid linje neoplasmer
vissa neoplasmer uttrycker markörer för både myeloid och lymfoid linjer.odifferentierad) eller nuvarande egenskaper hos båda linjerna (blandad fenotyp eller blandad linje akut leukemi).
tabell 3.Oms de las neoplasias mieloides y leucemias agudas
bibliograf Cuba
Bassan r, Hoelzer D. Modern terapi av akut lymfoblastisk leukemi. J Clin Oncol. 2011;29:523-43.
Burnett a, Wetzler M, L Jacobwenberg B. terapeutiska framsteg vid akut leukemi. J Clin Oncol. 2011;29:487-94. det är en av de mest kända. WHO-klassificeringen 2008 av lymfoida neoplasmer och bortom: utvecklande begrepp och praktiska tillämpningar. Blod. 2011 maj 12;117 (19): 5019-32. Epub 2011 Februari 7. Recension.
Cortes J, Hochhaus a, Hugues t, Kantajian H. frontlinjen och bärgning terapier med tyrosinkinashämmare och andra behandlingar vid kronisk Myeloid leukemi. J Clin Oncol. 2011;29:524-31.
Chin-hon Pui, Carroll WL, et al. Biologi, Riskstratifiering och terapi av akut leukemi hos barn: en uppdatering. J Clin Oncol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado Mr. Multicenter oberoende bedömning av vårkommer hos patienter med kronisk myeloid leukemi som behandlas med Imatinib. Journal National Cancer Institute. 2011;103:1-9.
Grever M, Lozanki G. Moderna strategier för hårig cell leukemi. J Clin Oncol. 2011:29;583-90.
Gribben JG, O ’ Brien S. uppdatering om Behandling av kronisk lymfocytisk leukemi. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Kronisk Myeloid Leukemi. Aktuella begrepp inom Fysiopatologi och behandling. Cancerolog Cuba. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib jämfört med imatinib / cytarabin för första linjens behandling av tidig Philadelphia-kromosompositiv kronisk Myeloid leukemi. Resultat av en randomiserad klinisk prövning av den mexikanska samarbetsgruppen leukemi. Klinisk Leukemi. 2008: 2(2);1128-32.
Lichtman MA. Klassificering och kliniska manifestationer av klonala myeloida störningar. Sv: Williams. Hematologi. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Molekylär Genetik av vuxen akut myeloid leukemi. Prognostiska och terapeutiska konsekvenser. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado m Mellado Y, Floresw RG, Pablo Vargas. Det finns många olika typer av produkter. Rev Fac med UNAM. 2010; 53:36-43.
Sanz M, Lo-Coco F. moderna metoder för behandling av akut promyelocytisk leukemi. J Clin Oncol. 2011;29:495-503.
Obs
* dysplasi. Det hänvisar till den cytomorfologiska förändringen som inkluderar dissociation av kärna-cytoplasmmognad (kom ihåg att kromatinmognad beror på DNA-och RNA-cytoplasma, därför stannar kärnan vid mognad medan cytoplasman fortsätter sin normala process) som producerar icke-livskraftiga celler och det finns intramedullär apoptos.