algorytm Basic Local Alignment Search Tool (BLAST) jest sercem bezpłatnego zestawu zasobów online dostępnych za pośrednictwem Narodowego Centrum Informacji biotechnologicznej (NCBI). Podczas gdy większość badaczy zdaje sobie sprawę z BLAST jako narzędzia do wyrównywania sekwencji, pakiet BLAST NCBI oferuje o wiele więcej! Omówię dogłębnie, jak wykorzystać te zasoby do zlokalizowania polimorfizmów pojedynczych nukleotydów (SNP) w genie; zaprojektować startery z Primer-BLAST; i zweryfikować cele starterowe.
Tip One: Jak znaleźć SNP
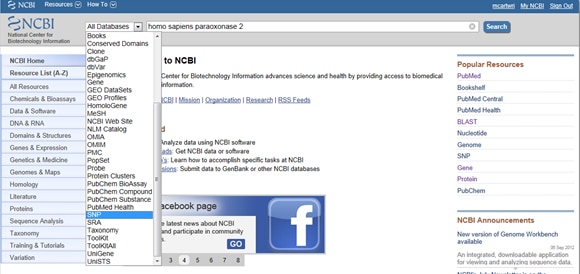
biorąc pod uwagę znaczenie SNP zarówno w chorobie, jak i badaniach, NCBI zapewnia narzędzia do zestawiania zgłaszanych SNP genu. Aby znaleźć SNP, zacznij od strony głównej NCBI i wpisz interesujący Cię gen w pasku wyszukiwania. Wybierz SNP z menu rozwijanego wszystkie bazy danych po lewej stronie paska wyszukiwania, Jak pokazano poniżej:
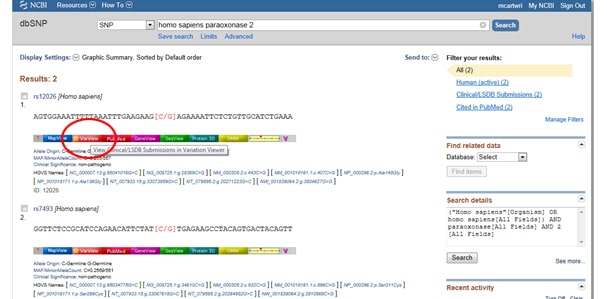
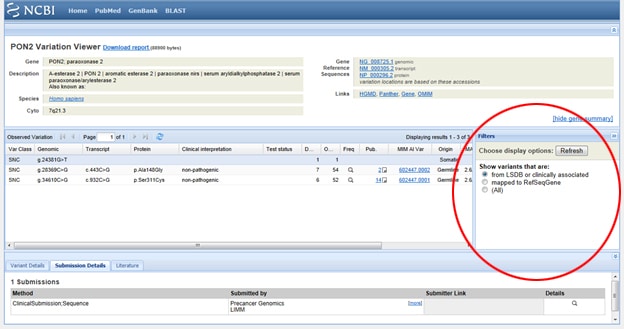
Trick pierwszy: trzeba filtrować wyniki tak, że patrzysz tylko na wyniki kliniczne? Przejdź do pola wyświetlania oznaczonego filtrami po prawej stronie listy SNP w obszarze obserwowane zmiany. Po wybraniu opcji filtrowania naciśnij przycisk Odśwież.
Wskazówka druga: jak zaprojektować podkłady
NCBI zapewnia Primer-BLAST do automatycznego projektowania podkładów na podstawie sekwencji zapytań. Aby rozpocząć projektowanie podkładów, przejdź do strony głównej BLAST i przewiń w dół do opcji Primer-BLAST w obszarze specialized BLAST. Wprowadź sekwencję docelową przez Wytnij i wklej lub, jeśli jest wymieniona w bazach danych NCBI, jako numer dostępu. Omówię kilka opcji dostosowywania poniżej, ale w tym momencie możesz generować podkłady bez dodatkowego dostosowywania!
Zakres: po prawej stronie pola do wprowadzania sekwencji, możesz określić dokładny zakres (ponumerowany od 5′ do 3′, Od początku sekwencji) celu, który będzie brany pod uwagę przy projektowaniu starterów do przodu i do tyłu.
Use my own forward primer (5′->3′ on plus strand): wybierz tę opcję, jeśli już zaprojektowałeś swoje Podkłady i chcesz, aby Primer-BLAST dostarczył analizy (np. Tm) na ich temat.
rozmiar produktu PCR: Ustaw zakres dopuszczalnych długości produktów PCR tutaj.
# starterów do zwrócenia: to ustawia preferowaną liczbę zestawów starterów do rozważenia. Zauważ, że nie jest to gwarancja, zwłaszcza jeśli twoje parametry są zbyt surowe lub bezsensowne(np. określiłeś produkt w rozmiarze produktu PCR, który nie może być większy niż 500 bp, ale w zakresie chcesz wziąć pod uwagę tylko podkłady oddalone od siebie o więcej niż 1 kb).
Temperatura topnienia podkładu: Pozwala to określić Tm (aby szybko odświeżyć temperaturę topnienia, zapoznaj się z naszymi poradami dotyczącymi qPCR i zwykłego projektu podkładu PCR).
Exon junction span: jeśli chcesz wykluczyć genomowe DNA (gdzie eksony są podzielone przez niekodujące introny), ustaw to na Starter musi rozciągać się na exon-exon junction.
sprawdzanie specyfiki: chyba, że chcesz, aby Primer-BLAST zwrócił podkłady, które pójdą poza cel (generalnie nie polecam!), pozostaw to zaznaczone i określ organizm, z którego pochodzą twoje próbki, a także jakiej bazy danych użyć, w zależności od tego, czy celujesz w mRNA, gDNA itp. Włączając kontrolę specyficzności, Primer-BLAST wykluczy startery, które mogłyby wzmocnić coś poza sekwencją docelową.
obsługa wariantu splicingu: jeśli wybierzesz tę opcję – wykonalną tylko wtedy, gdy pracujesz nad sekwencjami mRNA – Primer-BLAST Nie wykluczy par starterów, które mogłyby wzmocnić wiele wariantów splicingu mRNA Twojego celu. Nie oznacza to jednak, że da ci pary podkładów, które obejmują wszystkie znane warianty splicingu! Po prostu rozluźniasz swoje kryteria.
Po wprowadzeniu sekwencji i dostosowaniu w razie potrzeby przewiń w dół strony i po sprawdzeniu Użyj nowego widoku graficznego naciśnij Get Primers. Zwróci To mapę tego, gdzie sugerowane pary podkładu wzmocnią twój cel, a także analizę podkładów: ich długość, dokładna lokalizacja, odpowiednie Tm, GC% i wyniki odzwierciedlające samospełnienie (przy 0,00 nie odzwierciedlającym przewidywanego dopełnienia).
Wskazówka trzecia: jak przewidzieć cele podkładu
Jak sprawdzić, czy Twoje podkłady trafiły w coś poza celem? Idź do Primer-BLAST. W polu zapytania wprowadź swój podkład do przodu (od 5′ do 3′). Teraz wpisz 20 N z rzędu, aby rozdzielić startery na indywidualne, nie nakładające się wyrównania. Po znakach ” N „wprowadź swój odwrócony podkład (również od 5 'do 3′), Jak pokazano poniżej:
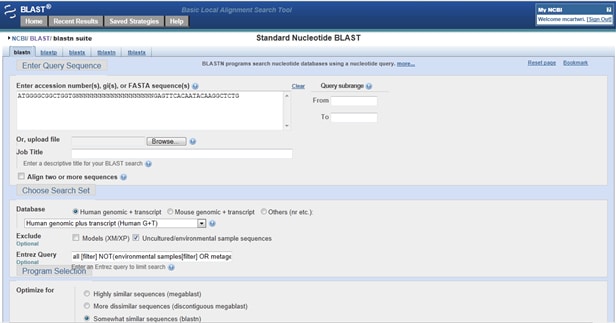
Po uzyskaniu wyników sprawdź je pod kątem pewnych kombinacji. Jeśli twój starter do przodu wyrównuje się na pasmie do przodu (pasem opatrzonym adnotacją Plus/Plus), a starter do tyłu wyrównuje się do tego samego uderzenia, ale na pasmie odwrotnym (pasem Plus/Minus), wtedy twoje startery mogą wzmocnić to uderzenie.
Trick Two: czy Twoje wyniki obejmują rzeczy, które prawdopodobnie nie zostały zanieczyszczone próbki PCR, takie jak pawiany oliwkowe i neandertalczycy? Jeśli pracujesz z próbkami ludzi lub myszy, upewnij się, że zostały one określone w obszarze baza danych. Alternatywnie można wykluczyć określone gatunki.
References and Additional Resources:
Blast Tips. 2007. NCBI. <http://www.ncbi.nlm.nih.gov/feed/rss.cgi?ChanKey=blasttips>
Frequently Asked Questions. NCBI BLAST Help. <http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=FAQ>
Madden T. The BLAST Sequence Analysis Tool. 2003. <http://www.ncbi.nlm.nih.gov/books/NBK21097/>
Mount DW. Using the Basic Local Alignment Search Tool. 2004. Cold Spring Harbor Protocols. <http://cshprotocols.cshlp.org/content/2007/7/pdb.top17.full>
Wheeler D and Bhagwat M. BLAST QuickStart. 2007. Humana Press Inc. <http://www.ncbi.nlm.nih.gov/books/NBK1734/>
czy to ci pomogło? Następnie podziel się z siecią.