artykuł przeglądowy
białaczka dla lekarza rodzinnego
białaczka dla lekarza ogólnego
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosk
szef Hematologii. Szpital Aniołów Del Pedregal. Meksyk, DF. e-mail: [email protected]
b medycyna wewnętrzna. Szpital Aniołów Del Pedregal. Meksyk. DF.
c medycyna wewnętrzna. Szpital Aniołów Del Pedregal. Meksyk. DF.
otrzymano: 17 października 2011
zaakceptowano: 07 stycznia 2012 r.
wprowadzenie
pomimo wielkich sukcesów, molekularnych i leczniczych badań białaczek, główne aspekty tego stanu nie są jeszcze wyraźnie znane, a lekarz nie jest hematologiem, za to, że celem tej pracy jest dostarczenie informacji, które studenci medycyny i lekarze w ogóle, i pozwala przede wszystkim zdobyć wiedzę na temat białaczek, ich terminowej diagnostyki i poszukiwania odniesień na wczesnym etapie hematolog.

definicja
białaczka jest terminem używanym do określenia grupy złośliwych zaburzeń krwi. Wczesna diagnoza jest konieczna, ponieważ pozwoli pacjentowi na wczesnym etapie udać się do lekarza hematologa, który przeprowadzi proces diagnostyczny i zaproponuje konkretne leczenie. Charakteryzuje się klonalną, autonomiczną i nieprawidłową proliferacją komórek, które dają początek pozostałym normalnym komórkom krwi (zachowanie guza jako całości).
powyższe implikuje, że wczesna komórka ulega zmianie genetycznej, która doprowadzi do niekontrolowanego nieprawidłowego klonu (Kolonii). Ta nieprawidłowa produkcja jest nieuporządkowana, ponieważ nieprawidłowe komórki rozmnażają się na obraz i podobieństwo, więc stopniowo zajmują przestrzeń normalnego szpiku kostnego i prowadzą do postępującej niedokrwistości, nieprawidłowego krwawienia i predyspozycji do infekcji. Z drugiej strony, gdy nieprawidłowe komórki wnikają w inne tkanki, to prowadzi do zaburzeń funkcjonowania danego organu, na przykład, infiltracja w ośrodkowy układ nerwowy w przypadku ostrej białaczki limfoblastycznej (LAL) może objawiać się bólem głowy, napady, skupiony zaburzeniami ruchu, wzrost ciśnienia śródczaszkowego, a niezdolność postawić wczesną diagnozę i zapewnić odpowiednie leczenie prowadzi do utraty funkcji i nieodwracalne skutki.
objawy kliniczne
obraz kliniczny jest zróżnicowany i będzie zależeć od rodzaju białaczki: ostrej lub przewlekłej, jednak dla wszystkich 2 istnieją niespecyficzne objawy kliniczne (występujące w każdej chorobie):
1. Zmęczenie.
2. Lekkie zmęczenie.
3. Uogólniona słabość.
4. Chęć pozostania w trybie snu lub w łóżku.
5. Wymaga czyjejś pomocy w zaspokojeniu twoich osobistych potrzeb.
białaczki przewlekłe są kursu leniwy i do 50% przypadków znaleziono na poprawkę klinikę lub zwykłe laboratorium ochotników, które są uważane za zdrowe i zwraca się do oddawania krwi, jednak, jak postępuje choroba, prezentowane są przejawami niewyjaśnione charakteru skargi, ale teraz są specyficzne (tab. 1).

w ostrych postaci, manifestacji, konkretnych wyniku niedoboru niektórych linii komórkowych:
1. Erytrocyty: zespół niedokrwistości, którego intensywność będzie zależeć od stopnia hipoksemii, niezależnie od stopnia niedokrwistości. Duszność od średniego wysiłku do ortopedii.
2. Płytki krwi: wybroczyny, wybroczyny w kończynach, aw cięższych, szeroko rozpowszechnionych przypadkach krwawienie suche i mokre z krwawieniem z nosa, krwotok dziąseł, krwiomocz, grzywa lub hematokezja. Bardzo ciężki w ośrodkowym układzie nerwowym (OUN).
3. Białe krwinki: gorączka, pocenie się, miejscowe infekcje do jawnej posocznicy (bakterie lub grzyby). Występują z neutropenią mniejszą niż 250 neutrofili / mm3.
zespół infiltrativo odnosi się do realizacji nieprawidłowego w dowolnej tkance, chociaż zazwyczaj jest to:
1. Powiększenie wątroby lub powiększenie śledziony (ryc. 5).
2. Adenomegalia (miejscowa lub uogólniona).
3. Cera białaczkowa.
4. Ból kości spowodowany rozszerzeniem szpiku kostnego.
5. Tkanki miękkie (mięsak granulocytowy).
6. Jądro.
7. SNC.
8. Dziąsła i dowolny węzeł (rys. 1).

zaburzenia metaboliczne: są wynikiem nieprawidłowej hiperprodukcji komórek złośliwych i zwiększonej apoptozy.
1. Kwasica.
2. Wzrost dehydrogenazy mlecznej (DHL).
3. Hiperkaliemia.
4. Hiperurykemia.
5. Wzrost mikroglobuliny β2

dane kliniczne dominuje jako kamień węgielny podejrzenie zdiagnozować białaczkę i każde cierpienie, ale to, co pozostaje-jest to dodatek do diagnostyki wspierany przez laboratorium kliniczne: a przenoszone z krwią, tak kompletne lub specjalne, co chcesz powiedzieć, dokładnych obserwacji rozmazów krwi obwodowej oraz personel techniczny przeszkolony w wykrywaniu nieprawidłowych komórek, a przede wszystkim białaczkowych.

zmiany laboratoryjne, które wymagają specjalnej kontroli obejmują:
1. Niedokrwistość (dowolnego stopnia).
2. Leukopenia lub leukocytoza (przewaga linii komórkowej).
3. Małopłytkowość.
4. Kombinacje: bikitopenia lub pancytopenia.
należy zachować szczególną ostrożność, gdy laboratorium zgłasza obecność nietypowych białych krwinek lub limfocytów (mogą to być blasty białaczkowe). Wskazane jest, aby poprosić eksperta o recenzję (ryc.2).

aspirator szpiku kostnego jest niezbędny do diagnozy (ryc.3), a 20% blastów jest wymagane do ustalenia kryterium ostrej białaczki w dowolnej jej odmianie.

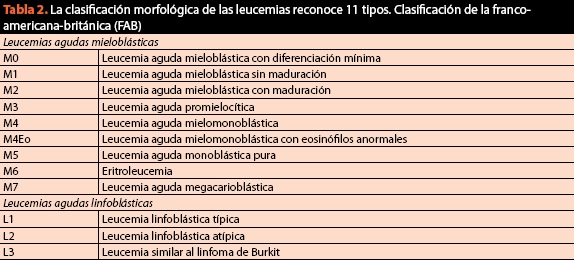
w ramach tej samej procedury konieczne będzie uzyskanie próbek do ostatecznej klasyfikacji cierpienia i zapytanie o kariotyp i immunofenotyp, ponieważ obecnie kryterium cytomorfologiczne jest niezbędne, ale już niewystarczające. W (Tabela 2) wymieniono główne klasyfikacje i obowiązujące w bieżącym złośliwych chorób krwi.
leczenie koncentruje się na 2 ważnych aspektach: pierwszym z nich jest specyficzny antileucémico i opiera się na stosowaniu leków pochodzenia chemicznego, które są znane pod nazwą chemioterapii, której głównym celem jest wyeliminowanie, czyli usunięcie wszystkich komórek białaczkowych organizmu. Drugim aspektem leczenia jest wsparcie powikłań, które zwykle występują u pacjentów przy przyjęciu.
leczenie koncentruje się na 2 ważnych aspektach: pierwszy z nich jest specyficzny antyleukemiczny i opiera się na stosowaniu leków pochodzenia chemicznego znanych pod nazwą chemioterapia, której głównym celem jest wyeliminowanie, czyli usunięcie wszystkich komórek białaczkowych z organizmu.
drugim aspektem leczenia jest wsparcie powikłań, które zwykle objawiają się u pacjentów przy przyjęciu:
1. Niedokrwistość.
2. Nieprawidłowe krwawienie.
3. Zakażenia płuc i uogólnione, między innymi (ryc. 4).
4. Wszelkie inne sąsiednie powikłania, które może mieć pacjent (współwystępowanie), takie jak wcześniej istniejące choroby, takie jak cukrzyca, nadciśnienie, choroby serca i inne częste choroby wśród pacjentów cierpiących na białaczkę.


dlatego bardzo ważne jest, aby pamiętać, że leczenie białaczki jest interdyscyplinarne, co wymaga zaangażowania innych specjalistów, aby wspierać hematolog.
leczenie antileucémico będzie również inne w przypadku różnych rodzajów białaczki i ostrych postaci. Jest on podzielony na 3 fazy:
1. Indukcja remisji. Celem jest osiągnięcie całkowitej remisji (CR), to znaczy normalizacji wartości krwi pacjenta, braku jakichkolwiek objawów lub oznak, że białaczka utrzymuje się wraz z naciekiem. Podczas tego procesu pacjent musi mieć” stan bez białaczki ” w szpiku kostnym, aw przyszłości musi nastąpić powrót do normalnej hematopoezy, a niestety w innych przypadkach dochodzi do siebie z chorobą, co sugeruje oporną białaczkę, której rokowanie jest kiepskie. Ten pierwszy proces może potrwać od 6 do 8 tygodni, aby osiągnąć RC.
2. Konsolidacja. Obejmuje stosowanie tych samych leków, które były stosowane w indukcji lub kombinacji innych chemioterapii, również w celu późniejszego zabicia resztkowych komórek złośliwych, które mogłyby rozwinąć oporność na pierwsze zastosowania.
3. Konserwacja. Lepiej jest utrzymywać pacjenta pod wpływem chemioterapii, gdy możliwe jest wystąpienie rodzącej się aktywności białaczki, a podczas leczenia utrzymać efekt do momentu ustąpienia choroby.

do tej pory dla ostrych postaci, coraz bardziej rygorystyczne kryteria do rozważenia RC stają się bardziej skomplikowane, więc lepiej jest szukać remisji molekularnej, która polega na znalezieniu zaburzenia chromosomalnego początkowej, a w przypadku białaczki promielocítica(M3-FAB) z t (15;17) początkowy, należy szukać na podstawie specyficznych badań molekularnych i cytogenetycznych, mimo że znajduje się w CR, ponieważ jeśli translacja jest zachowana, agresywne leczenie musi być kontynuowane aż do całkowitej eliminacji klonu. Ta odmiana białaczki powinna być uważana za potencjalnie uleczalną i jedną z najczęstszych w populacji Łacińskiej.
leczenie cierpienia będzie zależeć od usunięcia wszystkich istniejących komórek złośliwych od pacjenta. Ogólnie rzecz biorąc, niektóre białaczki mogą być podatne na wyleczenie tylko za pomocą chemioterapii, ale dziś dużą wagę należy przywiązywać do tak zwanych czynników prognostycznych, które opierają się na modelach matematycznych, które umożliwiają ocenę pacjentów w stopniu prognostycznym, jaki mają, i obejmują:
1. Rodzaj białaczki.
2. Początkowa zmiana molekularna i jej trwałość pomimo leczenia lub jego eradykacji.
3. wiek. Pacjenci w wieku powyżej 60 lat mają złe rokowanie w porównaniu z młodszymi pacjentami.
4. Chemioterapia. Konieczne jest stosowanie wskazanych leków, a przede wszystkim zalecanych dawek. Na przykład u dorosłych LAL stosowanie schematu HyperCyVAD (skalowane dawki cyklofosfamidu, winkrystyny, adriamycyny, deksametazonu w połączeniu z arabinozydem cytozyny i metotreksatem) Zapewnia 90% CR i leczy w 50% przypadków, dane wcześniej nie spotykane w innych schematach. To leczenie jest toksyczne i wymaga jego stosowania tylko w instytucjach, które mają wystarczające zasoby pomocnicze. Niestety, w naszym środowisku nie wszystkie ośrodki mają zalecane leki, a wyniki nie będą powtarzalne, ponieważ nie mają one również wystarczającego wsparcia pacjenta podczas fazy maksymalnej supresji szpiku.
5. Terapia wspomagająca. Osiągnięcia chemioterapii i nowych leków, nowych kombinacji i bardziej specyficzne obowiązki wymagają wdrożenia interdyscyplinarnych grup pod nadzorem hematologa; instalacji i użytkowania centralnych cewników; wsparcia banku krwi dla wsparcia transfuzji płytek krwi i krwinek czerwonych (w tym napromieniowane produkty); interwencji инфекциониста do wykrywania zakażeń; i właściwego wykorzystania antybiotyki lub leki przeciwgrzybicze, czy to profilaktyczne lub lecznicze, jeśli jest to konieczne; stworzenie pomieszczeń izolacyjnych z obsługą i usługami intendentury umożliwiającymi zapewnienie środowiska wolnego od bakterii, w tym sterylnego karmienia; laboratorium z protokołem przetwarzania próbek pacjentów według specjalnych protokołów leczenia i przetwarzanie specjalnych próbek (przygotowanie koncentratów leukocytów w cytometrii krwi buffy coat i osiągnięcie optymalnego odczytu); obszar przygotowania leków o wysokiej specjalizacji, a z drugiej strony najważniejszy jest personel pielęgniarski, pomocniczy i administracyjny, który z całą grupą oczekuje wyników, jakie mają w innych krajach.
6. Przeszczep szpiku kostnego (MO). Jest to złożony i kosztowny rodzaj leczenia, wymagający zgodnego dawcy Mo i nieaktywnego cierpienia z wysokim prawdopodobieństwem wczesnego lub późnego nawrotu lub ze słabymi czynnikami prognostycznymi. Jest to procedura o bardziej leczniczych tendencjach, ponieważ wykorzystuje megadozy chemioterapeutyczne do zabijania komórek białaczkowych, ale w próbie niszczy również normalne prekursory i konieczne staje się uzupełnienie nowego zgodnego normalnego szpiku kostnego. Rodzaj allogenicznego przeszczepu (brat zgodny identyczne) jest najlepszym wyborem, ponieważ odbiornik akceptuje nie tylko ze względu na tożsamość częściowo (unikalne przyjęcie wszystkich bliźniaków), a zatem występuje odrzucenie przeszczepu przeciwko gospodarzowi, co z kolei daje przeszczep przeciwko białaczce i zwiększa eliminację stempla leucémica, jednak w ciężkich przypadkach zespołu (stopień 3-4) częstość występowania wzrasta pomimo specyficznego zarządzania.
białaczki przewlekłe
1. Przewlekła białaczka limfocytowa. Występuje częściej u osób starszych, a kryterium jest utrzymywanie się limfocytozy powyżej 10 x 109 / l i mo z naciekaniem ponad 50% limfocytów o fenotypie CD5+. Kryterium leczenia jest podwojenie liczby limfocytów w ciągu jednego roku lub progresja adenomegalii lub powiększenia śledziony, chociaż niektóre przypadki unikają tego kryterium ze względu na obecność niedokrwistości hemolitycznej lub małopłytkowości autoimmunologicznej, a następnie wskazane jest leczenie oparte na kombinacji fludarabiny, cyklofosfamidu i prednizonu, w stadiach I I II obserwuje się tylko i oczekuje się ewolucji bez leczenia.
2. Przewlekła białaczka szpikowa (CML). W tej chorobie nastąpił znaczny postęp w wiedzy o obecności chromosomu Philadelphia, który został opisany w 1950 r. w tym mieście American Union i który na początku swojego istnienia oznaczał pierwszy marker chromosomowy w związku z nowotworami złośliwymi, jednak przez lata badań udało się poznać t (9;22)z funkcjonalną ekspresją chromosomu w celu wytworzenia onkoproteiny o wysokiej aktywności tyrokinokinazy, która zwiększa proliferację komórek, a z kolei wyjaśnia dużą leukocytozę i trombocytozę doświadczaną przez tych pacjentów, a także dużą powiększenie śledziony (ryc. 5).
do tych dat przypada 10-12 lat od odkrycia małej cząsteczki specjalnie skierowanej przeciwko temu cząsteczkowemu substratowi fosforanowemu dawcy w celu wewnętrznej regulacji komórki białaczkowej i jej substratów. Początkowo nazywany STI (signal transduction inhibitor) powodował konkurencyjne hamowanie fosforylacji, powodował apoptozę komórki, a pacjenci osiągali niespotykane wcześniej wyniki kliniczne z remisjami molekularnymi do 80-90% po 10 latach, drastycznie zmieniając naturalną historię choroby, która w poprzednim leczeniu wynosiła nie więcej niż 3 lata. Informacje te wprowadzają zmiany w Historii Naturalnej i prognozowaniu wcześniejszej śmiertelnej choroby w krótkim okresie.

te przykłady są podstawą postępu tak silne, że stało się w ostatnich latach, i że pożądane jest, aby wzbudzić zainteresowanie studentów medycyny, nauczycieli, ale przede wszystkim organów edukacyjnych, że białaczka ogólnie zajmuje pierwsze 5 miejsc częstość występowania złośliwych chorób dorosłych i pierwsze miejsca w dzieci, więc Hematologia, konieczne jest włączenie do podstawowych dyscyplin w programie nauczania medycyny i jeszcze jako uczestnika pomimo swojej prostoty brytyjska klasyfikacja francusko-amerykańska (FAB) może prowadzić do błędów diagnostycznych, a zatem terapeutycznych, do 20% przypadków. Z tego powodu klasyfikacja za pomocą metod immunohistochemicznych i biologii molekularnej stała się niezbędnym wymogiem dla prawidłowej klasyfikacji i późniejszego zarządzania pacjentami (ryc.6).

w przeciwieństwie do klasyfikacji FAB, who (tabl. 2) odzwierciedla zmianę paradygmatu, przez który rozumiemy choroby hemáticas, po raz pierwszy conjuntaron informacji genetycznej, cech morfologicznych, citoquímicas i inmunofenotípicas z badań klinicznych w algorytmach diagnostycznych nowotworów tkanki krwiotwórczej; względne znaczenie każdego kryterium różni się między nowotworami i nie ma „złotego standardu” dla klasyfikacji wszystkich złośliwych chorób hematologicznych. Celem było zidentyfikowanie obiektów, które mogą zostać rozpoznane przez patologów i mają znaczenie kliniczne. Od czasu jego wprowadzenia w 2001 r.wprowadzono różne zmiany w celu aktualizacji jego Zawartości w związku z najnowocześniejszymi odkryciami. Najnowszy przegląd pochodzi z 2008 r.i klasyfikuje nowotwory hematologiczne w następujący sposób:
1. Nowotwory szpikowe.
2. Nowotwory limfoidalne.
3. Choroby komórek tucznych lub komórek zagruntowanych
4. Histiocytarne i dendrytyczne choroby komórkowe

NEOPLASIS MIELOIDES
na końcu mieloides są dziedziczone od rodziców w szpiku kostnym, różnicują się w czerwone krwinki, granulocyty (neutrofile, bazofile i eozynofile), monocyty i megacariocitos. W klasyfikacji FAB uznawane są 3 główne kategorie:
1. Ostra białaczka szpikowa.
2. Zespoły mielodysplastyczne.
3. Nowotwory mieloproliferacyjne.
najważniejszymi determinantami uznanych kategorii są ich cechy morfologiczne, histochemiczne i immunofenotypowe oraz odsetek blastów, linia komórkowa i stopień różnicowania komórek nowotworowych (ryc.7). W ostatnich latach genetyki (citogenéticas i molekularnych), jak również wstępnego przetwarzania i ewolucji mielodisplasia, wykazały znaczący wpływ na zachowanie kliniczne takich chorób, które nie zawsze są odpowiednio skorelowane z kategorii FAB, więc dyskusja centralny do przeklasyfikowania było rozróżnienie patologicznych osób i czynników prognostycznych, w celu uzyskania kategorii o znaczeniu klinicznym i istotności dla patolog. Niektóre zmiany genetyczne wydają się definiować różne choroby, a inne są czynnikami rokowania danej choroby.

obecnie klasyfikacja who zbiera cierpienia mieloides na 4 główne grupy:
1. Choroby mieloproliferacyjne
2. Zespoły mielodysplastyczne
3. Choroby mielodysplastyczne / mieloproliferacyjne
4. Ostre białaczki szpikowe

choroby mieloproliferacyjne to grupa zaburzeń klonalnych związanych z proliferacją jednej lub więcej linii szpikowych. Staje się coraz bardziej oczywiste, że choroby te są często związane z mutacjami, które prowadzą do nieprawidłowego wzrostu aktywności kinaz tyrozynowych i proliferacji komórek progenitorowych szpiku kostnego, niezależnie od czynników wzrostu. Odsetek wybuchów w szpiku kostnym jest normalny lub nieznacznie podwyższony, ale zawsze jest mniejszy niż 20%. Hematopoeza jest zwykle skuteczna, co powoduje wzrost liczby jednej lub więcej dojrzałych komórek we krwi obwodowej. Prototypem nowotworów mieloproliferacyjnych jest przewlekła białaczka szpikowa z dodatnim chromosomem Philadelphia (Ph1) (BCR/ABL). Inne obiekty, które zostaną uwzględnione:
1. Policytemia Vera.
2. Idiopatyczne zwłóknienie szpiku.
3. Pierwotna pierwotna trombocytemia.
4. Przewlekła białaczka eozynofilowa.
5. Przewlekła białaczka neutrofilowa.
6. Mastocytoza.
7. Nieklasyfikowalne nowotwory mieloproliferacyjne.

zespoły mielodisplásicos odnoszą się do zaburzeń, które charakteryzują się produkcji komórek nieskuteczne i dysplazji, ze zmiennym ryzykiem transformacji w białaczkę. Komórkowość w szpiku kostnym jest często zwiększona, ale bardzo zmienna. Występuje dojrzewanie, ale także dysplazja jednej lub więcej linii szpikowych. Hematopoeza jest nieskuteczna i dlatego istnieją cytopenia. Ten artykuł zawiera:
1. Oporna na leczenie cytopenia z dysplazją * jednej linii.
• niedokrwistość oporna na leczenie.
• neutropenia refrakcyjna.
• małopłytkowość oporna.
2. Niedokrwistość oporna na leczenie z pierścieniowymi sideroblastami.
3. Oporna na leczenie cytopenia z dysplazją kilku linii.
4. Niedokrwistość oporna na leczenie z nadmiarem blastów.
5. Zespół mielodysplastyczny z d (5q).
6. Nieklasyfikowalny zespół mielodyspatyczny.
7. Młodzieńczy zespół mielodysplastyczny, obejmuje jednostkę czasową znaną jako cytopenia oporna na leczenie młodzieńcze.
zespoły mielodysplastyczne/mieloproliferacyjne obejmują zaburzenia, w których współistnieją cechy dysplastyczne i proliferacyjne. W tej grupie występuje białaczka młodzieńcza mielomonocytowa, która jest reprezentatywna dla obu zespołów (mielodysplastyczna i mieloproliferacyjna). Prawie połowa pacjentów ma normalne lub niskie liczby neutrofili i dysplazję kilku linii komórkowych bez organomegalii i szpiku kostnego z morfologią, która nasila się w kierunku opornej na leczenie niedokrwistości z nadmiarem blastów, ale z monocytozą. Inni pacjenci mają intensywną neutrofilię, monocytozę i powiększenie śledziony. Nie wiadomo jeszcze, czy są to 2 różne choroby, mielodysplastyczne i mieloproliferacyjne; jednak do tej pory nie ma różnic w nieprawidłowościach cytogenetycznych ani wzorcach wzrostu kolonii in vitro ani ich ewolucji klinicznej, dlatego istnieje sprzeczność między klinicystami a patologami w zależności od ich miejsca w klasyfikacji. Według najnowszej wersji w tym artykule znajdują się:
1. Przewlekła białaczka szpikowa.
2. Nietypowa przewlekła białaczka szpikowa(ujemna dla BCR / ABL).
3. Młodzieńcza białaczka mielomonocytowa.
4. Nieklasyfikowany zespół mielodysplastyczny / mieloproliferacyjny.
w kategorii białaczki, ostre mieloblásticas (LAM) (która jest określona w procentach 20% mieloblastyczny w szpiku kostnym lub w sagre obwodowej lub obecności nieprawidłowości, cytogenetyka, w szczególności pomimo liczby blastów) rozpoznaje następujące grupy:
1. LAM z nawracającymi translacjami cytogenetycznymi.
2. Lam o właściwościach mielodysplastycznych.
3. LAM i SMD związane z leczeniem przeciwnowotworowym.
4. Lam nie został sklasyfikowany.
5. Mięsak szpikowy.
6. Proliferacje szpikowe związane z zespołem Downa.
7. Plazmocytoidalny nowotwór blastyczny komórek dendrytycznych.
nowotwory limfoidalne
to te, które pochodzą z komórek, które normalnie rozwijają się w limfocyty T (cytotoksyczne, współpracujące lub regulatorowe LT) lub limfocyty B (limfocyty lub komórki plazmatyczne). Zasadniczo nowotwory limfoidalne dzielą się na te, które pochodzą z prekursorów limfoidalnych i te, które pochodzą z dojrzałych limfocytów i komórek plazmatycznych, a następnie grupują się zgodnie z ich kretem (B lub T).
historycznie nowotwory limfoidalne, które są obecne w szpiku kostnym i obejmują szpik kostny, zostały oddzielone od tych przedstawionych jako nowotwór (chłoniak). Jednak obecnie wiadomo, że każdy chłoniak może mieć kliniczne objawy białaczki i że każda białaczka może czasami objawiać się jako guz (mięsak granulocytów). W klasyfikacji WHO diagnoza kilku nowotworów limfoidalnych zależy nie tylko od anatomicznej lokalizacji komórek nowotworowych, ale także od morfologicznie określonego pochodzenia komórki nowotworowej. Rozważania te zniosły znaczenie terminów klasyfikacji FAB L1 i L2, ponieważ nie korelują one z ich immunofenotypem, nieprawidłowościami genetycznymi lub ich przebiegiem klinicznym(ryc.8). L3 jest równy Burkittowi w stadium leucémica i musi zostać zdiagnozowany jako taki.

1. Nowotwory prekursorów. Istnieje konsensus, że nowotwory progenitorowe, które są reprezentowane jako guzy lite i te związane ze szpikiem kostnym i krwią, są biologicznie tą samą chorobą o różnych objawach klinicznych. Większość nowotworów progenitorowych limfoidów jest reprezentowana jako białaczki, dlatego zdecydowano, że w klasyfikacji należy zachować termin LAL dla fazy białaczkowej nowotworów progenitorowych typu B I T. istnieją 2 główne kategorie:
* białaczki / chłoniaki progenitorowe B.
• białaczki/chłoniaki progenitorowe t.
2. Dojrzałe nowotwory komórek B. Proponowana klasyfikacja traktuje chłoniaki i białaczki tego samego typu komórek jako tę samą chorobę o różnych objawach klinicznych lub stadiach. Specyficzne choroby pochodzące z dojrzałych komórek B są następujące:
1. Przewlekła białaczka limfocytowa / chłoniak małych limfocytów.
2. Chłoniak limfoplazmatyczny.
3. Chłoniak z komórek płaszcza.
4. Białaczka prolimfocytowa komórek B
5. Chłoniak grudkowy.
6. Rozlany chłoniak wielkokomórkowy B.
• wewnątrznaczyniowy chłoniak wielkokomórkowy B.
• pierwotny chłoniak śródpiersia wielkokomórkowego B.
• chłoniak wielkokomórkowy B (związany z wirusem Epsteina Barr-EBV).
* bogaty w histiocyty chłoniak B wielkokomórkowy i limfocyty T
* rozlany chłoniak B wielkokomórkowy ośrodkowego układu nerwowego.
* pierwotny skórny chłoniak B o dużych komórkach.
• rozlany chłoniak z dużych komórek B u osób starszych, dodatni pod względem EBV.
• chłoniak pochmablastyczny.
• pierwotny chłoniak opłucnowy.
• chłoniak z dużych komórek B dodatni pod względem alkomy (ALK).
• Burkitta.
7. Chłoniak z komórek B strefy marginalnej.
8. Chłoniak z komórek B zewnątrznodalnej strefy brzeżnej.
9. Chłoniak z komórek B strefy brzeżnej śledziony
10. Białaczka komórkowa włochata.
11. Plazmocytoma / szpiczak komórek plazmatycznych.

na końcu linii MIELOIDES i limfoidalnych
w niektórych nowotworów, wyrażają zakładek jako linii mielodes jako limfoidalnych, to grupa reprezentuje białaczka niosących niejednoznaczne, że są to te, które LUB NIE mają możliwości linii linfoide lub szpikowej (białaczka niezróżnicowanych) lub stanowią możliwości dla obu rzędów (białaczka ostry mieszany fenotyp lub rzędów mieszanych).
Tabela 3.Clasificación de la OMS de las neoplassias mieloides y leucemias agudas
Bibliografia
Bassan R, Hoelzer D. nowoczesna terapia ostrej białaczki limfoblastycznej. J Clin Oncol. 2011;29:523-43.
Burnett a, Wetzler m, Löwenberg B. postępy terapeutyczne w ostrej białaczce. J Clin Oncol. 2011;29:487-94.
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Krew. 2011 May 12; 117 (19): 5019-32. Epub 2011 Feb 7 Przegląd.
Cortes J, Hochhaus a, Hugues T, Kantajian H. Front Line and Salvage Therapies with Tyrosine Kinase Inhibitors and other Treatments in Chronic Myeloid Leukemia. J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologia, stratyfikacja ryzyka i terapia ostrej białaczki dziecięcej: aktualizacja. J Clin Oncol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado Mr.Multicenter independent assessment of ourcomes in chronic myeloid leukemia patients treated with Imatinib. Journal National Cancer Institute. 2011;103:1-9.
Grever M., Łozanki G. Nowoczesne strategie białaczki włochatokomórkowej. J Clin Oncol. 2011:29;583-90.
Gribben JG, O ’ Brien S. Update on Therapy of Chronic Lymphocytic Leukemia. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Przewlekła Białaczka Szpikowa. Aktualne koncepcje w Fizjopatologii i leczeniu. Cancerología. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatynib porównywano z imatynibem / Cytarabiną w leczeniu pierwszego rzutu wczesnej przewlekłej białaczki szpikowej z chromosomem Philadelphia. Wyniki randomizowanego badania klinicznego meksykańskiej grupy białaczki. Białaczka Kliniczna. 2008: 2(2);1128-32.
Lichtman MA. Klasyfikacja i objawy kliniczne klonalnych zaburzeń mieloidalnych. En: Williams. Hematologia. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Molecular Genetics of adult acute myeloid Leukemia. Implikacje prognostyczne i terapeutyczne. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rev Fac Med UNAM. 2010; 53:36-43.
Sanz m, Lo-Coco F. nowoczesne podejścia do leczenia ostrej białaczki promielocytowej. J Clin Oncol. 2011;29:495-503.
Uwaga.
* dysplazji. Odnosi się to do zmiany cytomorfologicznej, która obejmuje dysocjację dojrzewania jądra-cytoplazmy (pamiętaj, że dojrzewanie chromatyny zależy od dna i cytoplazmy RNA, więc jądro zatrzymuje się w dojrzewaniu, a cytoplazma podąża za swoim normalnym procesem), co prowadzi do nierentownych komórek i apoptozy wewnątrzszpikowej.