kertausartikkeli
Leukemia-yleislääkärille
Leukemia yleislääkärille
Rafael Hurtado Monroyalle, Braulio Solano Estradabille, Pablo Vargas Viveroscille
hematologian osaston johtaja. Sairaala Ángeles del Pedregal. Meksiko, DF. Sähköpostiosoite: [email protected]
b Sisätaudit. Sairaala Ángeles del Pedregal. Meksiko. DF.
c Sisätaudit. Sairaala Ángeles del Pedregal. Meksiko. DF.
Received: October 17, 2011
Accepted: 07 tammikuu 2012
johdanto
huolimatta leukemiatutkimuksen suurista molekyylitason ja terapeuttisista edistysaskeleista, ei-hematologi ei vielä tunne tämän tilan perusnäkökohtia, joten tämän työn tavoitteena on tarjota perustavaa tietoa lääketieteen opiskelijoille ja lääkäreille yleensä, ja tämä mahdollistaa ennen kaikkea yleisen tiedon hankkimisen leukemioista, niiden oikea-aikaisesta diagnosoinnista ja varhaisen yhteyden etsimisen hematologiin.

määritelmä
Leukemia on termi, jolla määritellään ryhmä pahanlaatuisia verisairauksia. Varhainen diagnoosi on välttämätöntä, koska sen avulla potilas voi mennä aikaisin hematologian erikoislääkärin kanssa, joka johtaa diagnostista prosessia ja tarjoaa erityistä hoitoa. Sille on ominaista ottaa klooninen, autonominen ja epänormaali proliferaatio solujen, jotka aiheuttavat loput normaalien solujen veren (kasvaimen käyttäytymistä yleensä).
Tämä viittaa siihen, että varhainen solu käy läpi geneettisen muutoksen, joka aiheuttaa itsestään poikkeavan kloonin (yhdyskunnan) hallitsemattomasti. Tämä epänormaali tuotanto on häiriintynyt, koska epänormaali solut lisääntyvät kuvan ja kaltaiseksi itsestään, joten ne vähitellen miehittää tilaa normaalin luuytimen ja aiheuttaa progressiivista anemiaa, epänormaali verenvuoto ja alttius infektioille. Toisaalta, kun epänormaali solut tunkeutuvat muihin kudoksiin, on epäonnistuminen toiminnan elimen kyseessä, esimerkiksi tunkeutuminen keskushermostoon, joka esiintyy akuutti lymfoblastinen leukemia (LAL) voi ilmetä päänsärky, kouristukset, keskittynyt moottori muutoksia, lisääntynyt kallonsisäinen paine, ja epäonnistuminen tehdä varhainen diagnoosi ja tarjota asianmukaista hoitoa, esittää toiminnan menetys ja peruuttamattomia seurauksia.
kliiniset oireet
kliininen kuva on moninainen ja riippuu leukemiatyypistä: akuutista tai kroonisesta, mutta 2: lla on ei-spesifisiä kliinisiä ilmenemismuotoja (joita esiintyy missä tahansa sairaudessa):
1. Väsymys.
2. Helppo väsymys.
3. Yleinen heikkous.
4. Haluaa jäädä lepäämään tai sänkyyn.
5. Se vaatii jonkun apua täyttääkseen henkilökohtaiset tarpeesi.
krooniset leukemiat ovat velttoja, ja jopa 50% tapauksista havaitaan tavanomaisessa kliinisessä tai laboratoriokatsauksessa vapaaehtoisilla, joita pidetään terveinä ja jotka tulevat luovuttamaan verta.taudin edetessä on kuitenkin esitetty epäspesifisiä ilmenemismuotoja, mutta nyt ne ovat spesifisiä (Taulukko 1).

akuuteissa muodoissa spesifiset ilmenemismuodot johtuvat yhden solulinjan puutoksesta:
1. Erytrosyytit: aneeminen oireyhtymä, jonka voimakkuus riippuu hypoksemian asteesta riippumatta anemian asteesta. Keskipitkän rasituksen hengenahdistus, kunnes ortoprea.
2. Verihiutaleet: petekiat, ekkymoosi raajoissa, ja vakavammissa yleistyneissä tapauksissa, kuiva ja märkä verenvuoto, johon liittyy nenäverenvuoto, gingivorragia, hematuria, mane tai hematokesia. Erittäin vaikea keskushermostossa.
3. Leukosyytit: kuume, voimakas hikoilu, paikalliset infektiot jopa frank verenmyrkytys (bakteerit tai sienet). Niitä esiintyy neutropenian ollessa alle 250 neutrofiilien kokonaismäärää / mm3.
infiltratiivinen oireyhtymä: viittaa epänormaaliin implantaatioon missä tahansa kudoksessa, vaikka se on yleistä:
1. Hepatomegalia tai splenomegalia (kuva 5).
2. Adenomegalia (paikallinen tai yleistynyt).
3. Leukeeminen iho.
4. Luuytimen laajenemisesta johtuva luukipu.
5. Pehmytkudokset (granulosyyttinen sarkooma).
6. Kives.
7. SNC.
8. Kumit ja kaikki paikat (kuva 1).

metaboliset häiriöt: ne johtuvat pahanlaatuisten solujen epänormaalista liikatuotannosta ja lisääntyneestä apoptoosista.
1. Asidoosi.
2. Suurentunut maitohappodehydrogenaasi (LLD).
3. Hyperkalemia.
4. Hyperurikemia.
5. Lisääntynyt β2 mikroglobuliini.

tunnistamisessa epänormaaleja soluja, erityisesti leukemia.

erityistä tarkastelua vaativia laboratoriomuutoksia ovat:
1. Anemia (mikä tahansa luokka).
2. Leukopenia tai leukosytoosi (suurin osa solulinjasta).
3. Trombosytopenia.
4. Yhdistelmävalmisteet: bikitopenia tai pansytopenia.
erityistä varovaisuutta tulee noudattaa, kun laboratorio ilmoittaa leukosyyttien tai epätyypillisten lymfosyyttien esiintymisestä (voivat olla leukeemisia blasteja). On suositeltavaa pyytää asiantuntijan arvio (kuva 2).

Luuydinaspiraatio on välttämätön taudinmäärityksen kannalta (kuva 3), ja 20% blasteista tarvitaan akuutin leukemian kriteerien määrittämiseen jollekin sen lajeista.

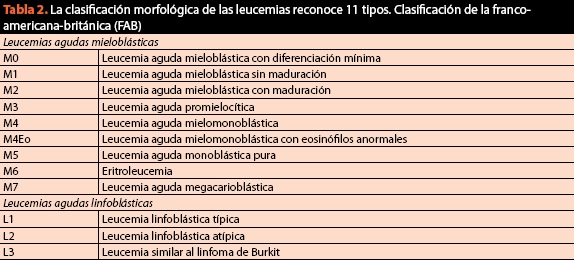
samalla menetelmällä on otettava näytteet tilan lopullista luokittelua ja pyyntöä varten karyotyyppi ja immunofenotyyppi, koska tällä hetkellä sytomorfologinen kriteeri on elintärkeä, mutta ei enää riittävä. Pahanlaatuisten verisairauksien nykyinen ja nykyinen luokitus on lueteltu taulukossa 2.
hoito tähtää kahteen tärkeään seikkaan: ensimmäinen niistä on spesifinen leukemialääke, ja se perustuu kemiallista alkuperää olevien lääkkeiden, niin sanottujen solunsalpaajien käyttöön, jonka päätavoite on hävittää eli hävittää kaikki leukemiasolut elimistöstä. Toinen osa hoitoa on tukea komplikaatioita, joita yleensä esiintyy potilailla ottamista.
hoito tähtää kahteen tärkeään seikkaan: ensimmäinen niistä on spesifinen leukemialääke, ja se perustuu kemiallista alkuperää olevien lääkkeiden, niin sanottujen solunsalpaajien, käyttöön, jonka päätavoite on hävittää eli poistaa kaikki leukemiasolut elimistöstä.
toinen osa hoitoa on sellaisten komplikaatioiden tukeminen, joita potilailla yleensä esiintyy sisäänpääsyssä, kuten:
1. Anemia.
2. Epänormaali verenvuoto.
3. Muun muassa keuhko-ja yleistyneet infektiot (Kuva 4).
4. Kaikki muut vierekkäiset komplikaatiot, joita potilaalla voi olla (samanaikainen sairastuvuus), kuten aiemmat tilat, kuten diabetes, verenpainetauti, sydänsairaus, ja muut sairaudet, jotka ovat yleisiä potilailla, jotka kärsivät leukemiasta.


siksi on erittäin tärkeää ottaa huomioon, että leukemian hoito on monitieteistä ja siihen osallistuu muita asiantuntijoita, kuten hematologin tuki.
Leukemialääkitys on myös erilainen eri leukemiatyypeissä ja akuuteissa muodoissa. Se jakautuu kolmeen vaiheeseen:
1. Remission induktio. Tavoitteena on saavuttaa täydellinen remissio (CR), eli potilaan veriarvojen normalisointi, sellaisten oireiden tai merkkien puuttuminen, että leukemia jatkuu infiltraatiolla. Prosessin aikana potilaalla pitäisi olla” leukemiaton tila ” luuytimessä ja tulevaisuudessa pitäisi olla toipuminen normaaliin hematopoieesiin, ja valitettavasti muissa tapauksissa he toipuvat taudin kanssa, joka puhuu resistentistä leukemiasta, jonka ennuste on kauhea. Tämä ensimmäinen prosessi voi kestää 6-8 viikkoa saavuttaa CR.
2. Yhdistäminen. Siinä käytetään samoja lääkkeitä, joita käytettiin muiden solunsalpaajien induktiossa tai yhdistelmässä, myös sellaisten jäljellä olevien pahanlaatuisten solujen hävittämiseksi, jotka voisivat kehittää resistenssin ensikäyttöä varten.
3. Huolto. On suotavaa, että potilas pysyy solunsalpaajahoidon vaikutuksen alaisena ennen alkavan leukemiavaikutuksen mahdollisuutta ja että hoito pitää vaikutuksen voimassa, kunnes tauti häviää.

toistaiseksi tiukimmat kriteerit CR: n huomioon ottamiseksi monimutkaistuvat, koska on parasta etsiä molekylaarinen remissio, johon kuuluu alkuperäisen kromosomimuutoksen etsiminen, kuten tapahtuu promyelosyyttisen leukemian (M3-FAB) tapauksessa T: llä (15;17) aluksi on etsittävä spesifisiä molekyyli-ja sytogeneettisiä tutkimuksia CR: stä huolimatta, koska jos translokaatio jatkuu, aggressiivista hoitoa on jatkettava kloonin täydelliseen eliminaatioon asti. Tämä erilaisia leukemia olisi pidettävä mahdollisesti parannettavissa ja on yksi yleisimmistä Latino väestöstä.
tämän jälkeen taudin paraneminen riippuu potilaan kaikkien olemassa olevien pahanlaatuisten solujen eliminoitumisesta. Yleensä jotkut leukemiat saattavat parantua pelkällä solunsalpaajahoidolla, mutta nykyään on kiinnitettävä paljon huomiota niin sanottuihin ennustetekijöihin, jotka perustuvat matemaattisiin malleihin, joiden avulla potilaat voidaan sijoittaa niiden ennusteasteeseen ja joita ovat:
1. Leukemian tyyppi.
2. Alkuperäinen molekyylimuutos ja sen pysyvyys hoidosta tai hävittämisestä huolimatta.
3. vuotiaana. Yli 60-vuotiaiden potilaiden ennuste on huono nuorempiin verrattuna.
4. Kemoterapia. On käytettävä ilmoitettuja lääkkeitä ja ennen kaikkea suositeltuja annoksia. Esimerkiksi aikuisilla LAL-potilailla Hyperkyvad-järjestelmän käyttö (suurentuneet annokset syklofosfamidia, vinkristiiniä, adriamysiiniä, deksametasonia yhdessä sytosiiniarabinosidin ja metotreksaatin kanssa) saavuttaa 90% CR: n ja parantaa 50%: ssa tapauksista, mitä ei ole aiemmin nähty muilla menetelmillä. Tämä käsittely on myrkyllistä ja edellyttää, että sitä käytetään vain laitoksissa, joilla on riittävät tukivarat. Valitettavasti ympäristössämme kaikilla keskuksilla ei ole suositeltuja lääkkeitä ja tulokset eivät ole toistettavissa, koska niillä ei ole riittävästi tukea potilaalle maksimaalisen myelosuppression vaiheessa.
5. Tukihoitoa. Kemoterapian ja uusien lääkkeiden saavutukset, uudet yhdistelmät ja spesifisemmät pakottavat monitieteisten ryhmien täytäntöönpanon hematologin osoitteella; keskuslaskimokatetrien asennus ja käyttö; veripankin tuki verihiutaleiden ja erytrosyyttien verensiirtojen tukemiseksi (jopa tuotteet stellaatti); infektologin puuttuminen infektioiden havaitsemiseen ja antibioottien tai sienilääkkeiden asianmukainen käyttö, joko profylaktinen tai terapeuttinen, tarvittaessa; sellaisten eristyshuoneiden tarjoaminen, joissa on huolto-ja Majoituspalvelut, joiden avulla voidaan saavuttaa bakteeriton ympäristö, mukaan lukien steriili ruokinta; laboratorio, jossa on potilasnäytteiden hallintaprotokolla erityisissä hoitoprotokollissa ja erityisnäytteiden hallinta (leukosyyttikonsentraattien valmistus buffy Coatin hematisessa sytometriassa ja optimaalisen lukeman saavuttaminen); pitkälle erikoistuneiden lääkkeiden valmistusalue ja toisaalta tärkein on hoitotyön, avustavan ja hallinnollisen henkilöstön, joka koko ryhmän kanssa odottaa tuloksia, joita heillä on muissa maissa.
6. Luuydinsiirto (engl. Se on monimutkainen ja kallis hoitomuoto, joka vaatii yhteensopivan MO-luovuttajan ja inaktiivisen tilan, jolla on suuri todennäköisyys varhaiseen tai myöhäiseen uusiutumiseen tai huonoon ennusteeseen. Se on menettely, jolla on enemmän parantavia taipumuksia, koska se käyttää kemoterapian megadoseja leukemisten solujen hävittämiseen, mutta yritys, se myös hävittää normaalit esiasteet ja uuden yhteensopivan normaalin luuytimen korvaaminen on välttämätöntä. Tyyppi allogeeninen (sisarusten yhteensopiva identtinen) on paras valinta, koska vastaanotin hyväksyy vain identiteetti osittainen (vain täydellisen hyväksymisen ovat identtiset kaksoset), ja siten johtaa hylkäämiseen käänteishyljintäsairaus, mikä puolestaan johtaa käänteishyljintäsairaus ja lisää hävittämistä kloonien leukemia, kuitenkin vaikeissa tapauksissa oireyhtymä (luokat 3-4) sairastuvuus ja kuolleisuus kasvaa huolimatta erityistä käsittelyä.
krooninen leukemia
1. Krooninen lymfaattinen leukemia. Sitä esiintyy useammin iäkkäillä ihmisillä, ja kriteerinä on yli 10 x 109/l: n lymfosytoosin pysyvyys ja MO, jossa yli 50% lymfosyyteistä on infiltroitunut CD5+ – fenotyypillä. Kriteeri hoidon on päällekkäisyyttä huomioon lymfosyyttien vuodessa tai etenemistä adenomegalia tai splenomegalia, vaikka joissakin tapauksissa tämän standardin läsnäolo hemolyyttinen anemia tai trombosytopenia, autoimmuunitauti, ja sitten osoitti hoito perustuu yhdistelmä fludarabiini, syklofosfamidi, ja prednisoni, vaiheissa I ja II vain somenter ja havaittu ja odotettu kehitys ilman hoitoa.
2. Krooninen myelooinen leukemia (KML). Tässä sairaudessa on paljon edistystä tiedossa Philadelphia-kromosomin läsnäolosta, joka kuvattiin vuonna 1950 Amerikan Unionin kaupungissa, ja että sen alku oli ensimmäinen merkkikromosomi yhdessä maligniteetin kanssa, mutta vuosien tutkimuksen aikana onnistuimme tuntemaan T(9;22) kromosomin toiminnallisen ilmentymisen kanssa onkoproteiinin tuottamisella, jolla on suuri tyroksinokinaasiaktiivisuus, mikä lisää solujen proliferaatiota ja mikä puolestaan selittää näiden potilaiden suuren leukosytoosin ja trombosytoosin sekä suuren splenomegalian (kuva 5).
tällä hetkellä on kulunut 10-12 vuotta siitä, kun löydettiin pieni molekyyli, joka on erityisesti suunnattu tätä fosfaatin luovuttavaa molekyylisubstraattia vastaan leukemisen solun ja sen substraattien sisäiseen säätelyyn. Alun perin nimeltään STI (signal transduction inhibitor) tuotti kompetitiivisen fosforylaation eston, johti solun apoptoosiin ja potilaat saavuttivat kliinisiä tuloksia, joita ei ole koskaan ennen nähty molekulaarisilla remissioilla, jotka ovat jopa 80-90% 10-vuotiaana, mikä muutti dramaattisesti taudin luonnollista historiaa, esikäsittely ei ollut yli 3 vuotta. Tämä tieto tekee muutoksen taudin luonnonhistoriaan ja ennusteeseen, joka aiemmin johti kuolemaan lyhyellä aikavälillä.

nämä esimerkit ovat perusta edistyksestä, joka on niin voimakasta, että se on tapahtunut viime vuosina ja että on suotavaa sallia herättää lääketieteen opiskelijoiden, heidän opettajiensa, mutta erityisesti koulutusviranomaisten kiinnostus leukemiaan yleensä, joka on ensimmäiset 5 paikkaa pahanlaatuisten sairauksien esiintymistiheydestä aikuisilla ja lapsilla, joten hematologia on sisällytettävä ydinaineisiin opetussuunnitelmaan lääketieteen uran ja jopa osana kursseja jatko-opintoihin erikoistuminen.
vaikka Brittiläinen Ranskalais-amerikkalainen luokitus (FAB) on yksinkertaisuutensa vuoksi hyödyllinen, se voi johtaa diagnostisiin ja siten myös terapeuttisiin virheisiin jopa 20 prosentissa tapauksista. Tästä syystä immunohistokemian ja molekyylibiologian menetelmiin perustuvasta luokituksesta on tullut ehdoton edellytys potilaiden asianmukaiselle luokittelulle ja myöhemmälle hoidolle (kuva 6).

toisin kuin luokitus FAB, WHO (taulukko 2) heijastaa paradigman muutosta, jonka kautta ymmärrämme veren sairauksia, koska ensimmäistä kertaa yhdistettiin geneettinen informaatio, morfologiset, sytokemialliset ja immunofenotyyppiset tiedot hematopoieettisen kudoksen kasvainten diagnostisissa algoritmeissa havaittuihin kliinisiin löydöksiin; kunkin kriteerin suhteellinen merkitys vaihtelee kasvaimien välillä, eikä kaikkien hematologisten maligniteettien luokittelulle ole olemassa ”kultastandardia”. Tavoitteena oli määritellä entiteettejä, jotka patologit voisivat tunnistaa ja joilla olisi kliinistä merkitystä. Sen ilmestymisestä vuonna 2001 lähtien sen sisältöä on muutettu eri tavoin suhteessa ajankohtaisimpiin löytöihin. Viimeisin katsaus on vuodelta 2008 ja se luokittelee hematologiset maligniteetit seuraavasti:
1. Myelooiset kasvaimet.
2. Lymfakasvaimet.
3. Syöttösolu-tai rasvasolusairaudet.
4. Histiocytic ja dendritic solujen sairaudet.

MYELOOISET kasvaimet myeloidinen
myelooiset kasvaimet ovat peräisin luuytimen kantasoluista, erilaistuvat punasoluiksi, granulosyyteiksi (neutrofiilit, basofiilit ja eosinofiilit), monosyyteiksi ja megakaryosyyteiksi. FAB-luokitus tunnustaa 3 pääluokkaa:
1. Akuutti myelooinen leukemia.
2. Myelodysplastiset oireyhtymät.
3. Myeloproliferatiiviset kasvaimet.
morfologisten, histokemiallisten ja immunofenotyyppisten tunnistettujen luokkien tärkeimmät taustatekijät sekä blastisolujen prosenttiosuus, solulinja ja neoplastisten solujen erilaistumisaste (kuva 7). Viime vuosina geneettiset ominaisuudet (sytogeneettiset ja molekulaariset) sekä myelodysplasian esihoito ja kehitys osoittivat merkittävää vaikutusta näiden kärsimysten kliiniseen käyttäytymiseen, jotka eivät aina korreloi hyvin FAB: n kategorioiden kanssa. Jotkut geneettiset poikkeavuudet näyttävät määrittelevän eri sairauksia, kun taas toiset edustavat ennustavia tekijöitä tietyn sairauden.

tällä hetkellä WHO luokittelee myelooiset tilat neljään pääryhmään:
1. Myeloproliferatiiviset sairaudet
2. Myelodysplastiset oireyhtymät
3. Myelodysplastiset / Myeloproliferatiiviset sairaudet
4. Akuutit myelooiset leukemiat

Myeloproliferatiiviset sairaudet ovat ryhmä kloonisia sairauksia, jotka liittyvät yhden tai useamman myelooisen linjan proliferaatioon. On yhä selvempää, että näihin sairauksiin liittyy usein mutaatioita, jotka aiheuttavat poikkeavaa tyrosiinikinaasiaktiivisuuden lisääntymistä ja kasvutekijöistä riippumatonta progenitorisolujen proliferaatiota luuytimessä. Blastien osuus luuytimessä on normaali tai hieman korkea, mutta aina alle 20%. Hematopoieesi on yleensä tehokas, mikä johtaa yhden tai useamman kypsän solun määrän lisääntymiseen ääreisveressä. Myeloproliferatiivisten kasvainten prototyyppi on Philadelphia-kromosomipositiivinen (Ph1) krooninen myelooinen leukemia (BCR/ABL). Muut mukana olevat yhteisöt ovat:
1. Polysytemia vera.
2. Idiopaattinen myelofibroosi.
3. Primaarinen essentiaalinen trombosytemia.
4. Krooninen eosinofiilinen leukemia.
5. Krooninen neutrofiilinen leukemia.
6. Mastosytoosi.
7. Luokittelemattomat Myeloproliferatiiviset kasvaimet.

Myelodysplastisilla oireyhtymillä tarkoitetaan häiriöitä, joille on ominaista tehoton solutuotanto ja dysplasia, ja joilla on vaihteleva riski muuntua akuutiksi leukemiaksi. Sellulaarisuus luuytimessä on usein lisääntynyt, mutta hyvin vaihteleva. On kypsymistä, mutta myös dysplasia yhden tai useamman myelooisen linjan. Hematopoieesi ei tehoa ja siksi sytopenioita on olemassa. Tämä erä sisältää:
1. Tulenkestävä sytopenia, johon liittyy yksirivinen dysplasia*.
• Refraktaarinen anemia.
• Refraktaarinen neutropenia.
• Refraktäärinen trombosytopenia.
2. Tulenkestävä anemia rengasmaisilla sideroblasteilla.
3. Refraktaarinen sytopenia, johon liittyy useiden sukusolujen dysplasiaa.
4. Tulenkestävä anemia ylimääräisillä räjäytyksillä.
5. Myelodysplastinen oireyhtymä, johon liittyy d (5q).
6. Luokittelematon myelodyspastinen oireyhtymä.
7. Juveniili myelodysplastinen oireyhtymä, sisältää väliaikaisen kokonaisuuden, joka tunnetaan nimellä juvenile refraktäärinen sytopenia.
Myelodysplastisia / myeloproliferatiivisia oireyhtymiä ovat häiriöt, joissa esiintyy samanaikaisesti dysplastisia ja proliferatiivisia ominaisuuksia. Tähän ryhmään kuuluu myelomonosyyttinen juveniili leukemia, joka edustaa molempia oireyhtymiä (myelodysplastinen ja myeloproliferatiivinen). Lähes puolet potilaista esiintyy normaali tai alhainen neutrofiilien määrä ja dysplasia useiden solulinjojen ilman organomegalia ja luuytimen morfologia muistuttaa tulenkestävää anemiaa ylimääräinen blasteja, mutta monosytoosi. Muilla potilailla on vaikea neutrofiilia, monosytoosia ja splenomegaliaa. Ei ole vielä tiedossa, jos ne ovat 2 eri sairauksia, myelodysplastinen ja myeloproliferatiivinen; toistaiseksi ei kuitenkaan ole eroja sytogeneettisissä poikkeavuuksissa tai in vitro-pesäkkeiden kasvutavoissa tai niiden kliinisessä evoluutiossa, joten kliinikkojen ja patologien välillä on kiistaa sen mukaan, mikä on niiden paikka luokituksessa. Viimeisen version mukaan tähän luokkaan sijoittuvat:
1. Krooninen myelomonosyyttinen leukemia.
2. Epätyypillinen krooninen myelooinen leukemia (BCR / ABL-negatiivinen).
3. Juveniili myelomonosyyttinen leukemia.
4. Myelodysplastinen / myeloproliferatiivinen oireyhtymä, jota ei voida luokitella.
akuuttien leukemioiden mieloblásticas (LAM) – ryhmässä (joka määritellään yli 20%: n myeloblastien prosenttiosuutena luuytimessä tai verenohennusveren ääreisosassa tai erityisesti sytogeneettisen poikkeavuuden perusteella, huolimatta blastien esiintymisestä) tunnistaa seuraavat ryhmät:
1. LAM ja toistuvat sytogeneettiset translokaatiot.
2. LAM myelodysplastisilla ominaisuuksilla.
3. LAM ja MDS liittyvät antineoplastiset hoidot.
4. Minua ei voi luokitella.
5. Myelooinen sarkooma.
6. Myelooinen proliferaatio, joka liittyy Downin oireyhtymään.
7. Dendriittisolujen plasmasitoidiblastinen kasvain.
IMUKUDOSKASVAIMET
ovat sellaisia, jotka ovat peräisin soluista, jotka normaalisti kehittyvät T-lymfosyyteiksi (sytotoksisiksi LT-lymfosyyteiksi, niiden yhteistyökumppaneiksi tai säätelijöiksi) tai B-lymfosyyteiksi (lymfosyyteiksi tai plasmasoluiksi). Yleensä imukudoskasvaimet jaetaan lymfoidien esiasteista johdettuihin ja kypsistä lymfosyyteistä ja plasmasoluista saatuihin kasvaimiin, ja ne ryhmitellään tämän jälkeen sukujuuriensa (B tai T) mukaan.
historiallisesti luuytimessä esiintyvät lymfoomat on erotettu kasvaimina esiintyvistä lymfoomista (lymfooma). Kuitenkin, se on nyt tiedossa, että mikä tahansa lymfooma voi esittää kliinisiä piirteitä leukemia ja että mikä tahansa leukemia voi joskus esittää kasvain (granulosyyttinen sarkooma). WHO: n luokituksessa useiden lymfakasvainten diagnosointi riippuu paitsi kasvainsolujen anatomisesta sijainnista, myös kasvainsolun morfologisesti määritellystä alkuperästä. Nämä seikat tekivät tyhjäksi FAB-luokituksen termien L1 ja L2 merkityksellisyyden, koska ne eivät korreloi immunofenotyyppinsä, geneettisten poikkeavuuksiensa tai kliinisen kulkunsa kanssa (kuva 8). L3 vastaa leukemiavaiheessa Burkittin lymfoomaa ja se tulisi diagnosoida sellaiseksi.

1. Esiastekasvaimet. On yksimielisyys siitä, että prekursoriset kasvaimet, jotka esiintyvät kiinteinä kasvaimina, ja ne, joihin liittyy luuydintä ja verta, ovat biologisesti sama sairaus, jolla on erilaiset kliiniset esitykset. Useimmat prekursoriset imukudoskasvaimet esitetään leukemioina, joten sovittiin, että luokituksessa tulisi säilyttää termi Lal tyypin B ja T prekursoristen kasvainten leukemiavaihetta varten. On olemassa 2 pääluokkaa:
• Esiasteiset B-leukemiat / lymfoomat.
• Prekursorileukemia/lymfoomat T.
2. Kypsät B-solujen kasvaimet. Ehdotetussa luokittelussa lymfoomat ja leukemiat, jotka ovat samaa solutyyppiä, katsotaan samaksi sairaudeksi, jolla on erilainen kliininen esiintyminen tai vaihe. Spesifisiä sukukypsistä B-soluista johdettuja tauteja ovat seuraavat:
1. Krooninen lymfaattinen leukemia / pieni lymfosyyttilymfooma.
2. Lymfoplasmasyyttinen lymfooma.
3. Manttelisolulymfooma.
4. Prolymfosyyttinen B-soluleukemia.
5. Follikulaarinen lymfooma.
6. Diffuusi suurisoluinen lymfooma B.
• intravaskulaarinen suurisoluinen lymfooma B.
• primaarinen mediastinaalinen suurisoluinen lymfooma B.
• suurisoluinen lymfooma B (liittyy Epstein Barr virus-EBV: hen).
•• Histiosyytti-ja T-solurikas suuri B-solulymfooma.
* diffuusi keskushermoston suuri B-solulymfooma.
• * diffuusi primaarinen ihon suurisoluinen B-solulymfooma.
• iäkkäiden potilaiden diffuusi suuri B-solulymfooma, joka on positiivinen EBV: lle.
• pasmablastinen lymfooma.
• primaarinen pleuralymfooma.
• * Alkoma-positiivinen (ALK) suurisolulymfooma.
• Burkittin lymfooma.
7. Marginaalinen B-solulymfooma.
8. Ekstranodaalinen marginaalinen B-solulymfooma.
9. Pernan reunavyöhykkeen B-solulymfooma
10. Karvasoluleukemia.
11. Plasmosytooma / plasmasolujen myelooma.

myelooisten ja lymfaattisten linjojen kasvaimet
jotkut kasvaimet ilmentävät sekä myelooisten että lymfaattisten linjojen markkereita.erilaistumaton) tai molempien linjojen nykyiset ominaisuudet (sekafenotyyppi tai sekalinja akuutti leukemia).
Taulukko 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍA
Bassan R, Hoelzer D. akuutin lymfoblastisen leukemian moderni hoito. J Clin Onkol. 2011;29:523-43.
Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in Acute Leukemia. J Clin Onkol. 2011;29:487-94.
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 who classification of lymphoid neoplases and beyond: evolving concepts and practical applications. Verenkierto. 2011 May 12;117(19):5019-32. Epub 2011 Helmi 7. Arvostelu.
Cortes J, Hochhaus A, Hugues T, Kantajian H. Front Line-ja Pelastushoidot tyrosiinikinaasin estäjillä ja muut kroonisen myelooisen leukemian hoidot. J Clin Onkol. 2011;29:524-31.
Chin-Hon Pui, Carroll WL, et al. Biologia, Risk Stratification, ja hoito lasten akuutti Leukemia: päivitys. J Clin Onkol. 2011;29:551-65.
Gambacorti PC, Antolin L, Hurtado Mr. Multicenter independent assessment of ourcomes in chronic myelooinen leukemia patients treated with imatinibi. Journal National Cancer Institute. 2011;103:1-9.
Grever M, Lozanki G. Moderni strategioita Karvasoluleukemia. J Clin Onkol. 2011:29;583-90.
Gribben JG, O ’ Brien S. Update on Therapy of Chronic Lymphocytic Leukemia. J Clin Onkol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Krooninen Myelooinen Leukemia. Fysiopatologian ja hoidon nykyiset käsitteet. Cancerología. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinibia verrattiin Imatinibiin / Sytarabiiniin varhaisen Philadelphia-kromosomipositiivisen kroonisen myelooisen leukemian ensilinjan hoidossa. Tulokset satunnaistetusta kliinisestä tutkimuksesta meksikolaiselle Leukemiaryhmälle. Kliininen Leukemia. 2008: 2(2);1128-32.
Lichtman MA. Kloonisten myelooisten häiriöiden luokittelu ja kliiniset oireet. Fi: Williams. Hematologia. Mc Graw-Hill; 2010.
Marcucci G, Haferlach T, Dohner H. Molecular Genetics of adult acute myelooinen Leukemia. Prognostisia ja terapeuttisia vaikutuksia. J Clin Onkol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rev FAC Med UNAM. 2010; 53:36-43.
Sanz M, Lo-Coco F. Modern Approaches to treating Acute Promyelocytic Leukemia. J Clin Onkol. 2011;29:495-503.
Huomautus
* Dysplasia. Se viittaa sytomorfologiseen muutokseen, joka sisältää tuman ja sytoplasman kypsymisen dissosiaation (muista, että kromatiinin kypsyminen riippuu DNA: n ja RNA: n sytoplasmasta, joten Tuma pysähtyy kypsymiseen, kun sytoplasma jatkaa normaalia prosessiaan), joka tuottaa elinkyvyttömiä soluja ja on intramedullaarinen apoptoosi.