gennemgå artikel
leukæmi-for den praktiserende læge
leukæmi for den praktiserende læge
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
leder af afdelingen for Hæmatologi. Sygehus løjtnant del Pedregal. København, DF. E-mail-adresse: [email protected]
B Intern Medicin. Sygehus løjtnant del Pedregal. Mexico. DF.
C Intern Medicin. Sygehus løjtnant del Pedregal. Mexico. DF.
modtaget: 17. oktober 2011
accepteret: 07. januar 2012
introduktion
På trods af de store molekylære og terapeutiske fremskridt i studiet af leukæmier er de grundlæggende aspekter af denne tilstand endnu ikke klart kendt af ikke-hæmatologen, så formålet med dette arbejde er at give grundlæggende information til medicinstuderende og læger generelt, og det giver frem for alt mulighed for at opnå generel viden om leukæmier, deres rettidige diagnose og søge tidlig henvisning til hæmatologen.

DEFINITION
leukæmi er det udtryk, der bruges til at definere en gruppe maligne blodsygdomme. Tidlig diagnose er vigtig, da det giver patienten mulighed for at gå tidligt med hæmatologispecialisten, som vil lede diagnoseprocessen og tilbyde den specifikke behandling. Det er kendetegnet ved at have en klonal, autonom og unormal proliferation af cellerne, der giver anledning til resten af de normale celler i blodet (tumoradfærd generelt).
Dette indebærer, at en tidlig celle gennemgår en genetisk ændring, der vil medføre, at en unormal klon (koloni) i sig selv forekommer ukontrollabelt. Denne unormale produktion forstyrres, fordi de unormale celler formerer sig i billede og lighed af sig selv, så de gradvist optager rummet i det normale knoglemarv og forårsager progressiv anæmi, unormal blødning og disponering for infektioner. På den anden side, når unormale celler invaderer andre væv, vil der være svigt i det pågældende organs funktion, for eksempel infiltration til centralnervesystemet, der forekommer ved akut lymfoblastisk leukæmi (LAL) kan manifestere sig med hovedpine, anfald, fokuserede motoriske ændringer, øget intrakranielt tryk og manglende evne til at stille en tidlig diagnose og give tilstrækkelig behandling, vil medføre tab af funktion og irreversible konsekvenser.
kliniske manifestationer
det kliniske billede er forskelligt og afhænger af typen af leukæmi: akut eller kronisk, men for 2 er der ikke-specifikke kliniske manifestationer (som forekommer i enhver sygdom):
1. Træthed.
2. Let træthed.
3. Generel svaghed.
4. Ønsker at forblive hvile eller i sengen.
5. Det kræver hjælp fra nogen til at imødekomme dine personlige behov.
kroniske leukæmier er indolente, og op til 50% af tilfældene opdages i en rutinemæssig klinisk eller laboratorieanmeldelse hos frivillige, der betragtes som sunde og kommer til at donere blod, men efterhånden som sygdommen skrider frem, præsenteres uspecifikke manifestationer, men nu er de specifikke (tabel 1).

i akutte former er specifikke manifestationer afledt af mangel på en af cellelinjerne:
1. Erythrocytter: anemisk syndrom, hvis intensitet afhænger af graden af hypoksæmi uanset graden af anæmi. Dyspnø af medium anstrengelse indtil orthoprea.
2. Blodplader: petechiae, ekkymose i ekstremiteter og i mere alvorlige generaliserede tilfælde, tør og våd blødning med næseblod, gingivorrhagi, hæmaturi, manke eller hæmatokesi. Meget alvorlig i centralnervesystemet (CNS).
3. Leukocytter: feber, diaphorese, lokaliserede infektioner op til ærlig septikæmi (bakterier eller svampe). De forekommer med neutropeni mindre end 250 totale neutrofiler/mm3.
infiltrativt syndrom: henviser til unormal implantation i ethvert væv, selvom det er almindeligt:
1. Hepatomegali eller splenomegali (figur 5).
2. Adenomegali (lokal eller generaliseret).
3. Leukæmisk hud.
4. Knoglesmerter fra knoglemarvsudvidelse.
5. Blødt væv (granulocytisk sarkom).
6. Testikel.
7. SNC.
8. Tandkød og ethvert sted (figur 1).

metaboliske lidelser: de skyldes unormal hyperproduktion af maligne celler og øget apoptose.
1. Acidose.
2. Øget mælkesyrehydrogenase (LLD).
3. Hyperkalæmi.
4. Hyperurikæmi.
5. Øget mikroglobulin til mikroglobulin 2.

det kliniske bevis dominerer som hjørnestenen i de mistænkte diagnoser af akutte leukæmier og af enhver tilstand, men det følgende er at supplere diagnosen med støtte fra det kliniske laboratorium i cytometry komplet blodtælling, eller specielt, hvilket vil sige den grundige observation af det perifere blodudstrygning af det tekniske personale, der har den fulde forberedelse til identifikation af unormale celler, især leukæmi.

laboratorieændringer, der kræver særlig gennemgang, inkluderer:
1. Anæmi (enhver klasse).
2. Leukopeni eller leukocytose (overvejelse af en cellelinie).
3. Trombocytopeni.
4. Kombinationer: bicitopeni eller pancytopeni.
Der skal udvises særlig forsigtighed, når laboratoriet rapporterer tilstedeværelsen af leukocytter eller atypiske lymfocytter (kan være leukæmiske Blaster). Det anbefales at anmode om en ekspertanmeldelse (figur 2).

Knoglemarvsaspiration er afgørende for diagnosen (figur 3), og 20% af blaster er nødvendige for at fastlægge kriterierne for akut leukæmi i nogen af dens sorter.

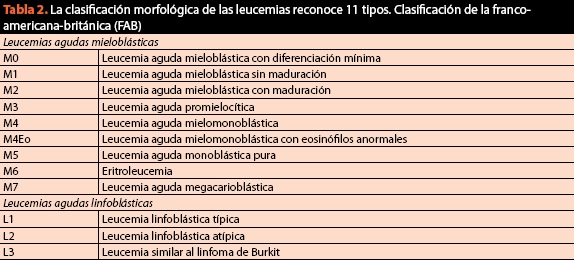
i samme procedure skal der udtages prøver til den endelige klassificering af tilstanden og anmodningskaryotype og immunofenotype, da det cytomorfologiske kriterium i øjeblikket er af vital betydning, men ikke længere er tilstrækkeligt. Den nuværende og nuværende klassificering af ondartede blodsygdomme er anført i tabel 2.
behandlingen er rettet mod 2 vigtige aspekter: den første er den specifikke antileukemiske og er baseret på brugen af stoffer af kemisk oprindelse, der er kendt som kemoterapi, hvis hovedmål er at udrydde, det vil sige eliminere alle leukæmiske celler fra kroppen. Det andet aspekt af behandlingen er støtte til komplikationer, der normalt forekommer hos patienter ved indlæggelse.
behandlingen er rettet mod 2 vigtige aspekter: den første er den specifikke antileukemiske og er baseret på brugen af stoffer af kemisk oprindelse, der er kendt som kemoterapi, hvis hovedmål er at udrydde, det vil sige eliminere alle leukæmiske celler fra kroppen.
det andet aspekt af behandlingen er støtten til de komplikationer, som patienter normalt præsenterer ved optagelse, såsom:
1. Anemia.
2. Unormal blødning.
3. Pulmonale og generaliserede infektioner, blandt andre (figur 4).
4. Eventuelle andre tilstødende komplikationer, som patienten kan have (co-morbiditet), såsom allerede eksisterende tilstande, f.eks. diabetes, hypertension, hjertesygdom og andre sygdomme, der er almindelige blandt patienter, der lider af leukæmi.


derfor er det meget vigtigt at tage højde for, at behandlingen mod leukæmi er tværfaglig, der involverer deltagelse af andre specialister som støtte til hæmatologen.
Antileukemisk behandling vil også være forskellig for forskellige typer leukæmi og for akutte former. Det er opdelt i 3 faser:
1. Induktion af remission. Målet er at opnå fuldstændig remission (CR), det vil sige normalisering af patientens blodværdier, fraværet af symptomer eller tegn på, at leukæmi fortsætter med infiltration. Under processen skal patienten have en” leukæmi-fri tilstand ” i knoglemarven, og fremtiden skal være genoprettelsen til en normal hæmatopoiesis, og desværre i andre tilfælde genvinder de med sygdommen, som taler om resistent leukæmi, hvis prognose er forfærdelig. Denne første proces kan tage 6 til 8 uger at opnå CR.
2. Konsolidering. Det involverer brugen af de samme lægemidler, der blev brugt til induktion eller kombination af andre kemoterapeutika, også med det formål at følge udryddelsen af resterende maligne celler, der kunne udvikle resistens over for første brug.
3. Vedligeholdelse. Det foretrækkes at holde patienten under virkningen af kemoterapi inden muligheden for begyndende leukæmisk aktivitet, og at den med behandlingen opretholder virkning, indtil sygdommen forsvinder.

indtil videre for akutte former bliver de strengeste kriterier for at overveje CR mere komplekse, da det er bedst at kigge efter molekylær remission, som involverer søgningen efter den indledende kromosomale ændring, som forekommer i tilfælde af promyelocytisk leukæmi (M3-FAB) med t (15;17) indledende skal det søges efter specifikke molekylære og cytogenetiske undersøgelser på trods af at de er i CR, da hvis translokationen fortsætter, skal den aggressive behandling fortsættes indtil den totale eliminering af klonen. Denne række leukæmi bør betragtes som potentielt hærdelig og er en af de mest almindelige i Latino-befolkningen.
helbredelsen af sygdommen afhænger derefter af eliminering af alle eksisterende maligne celler i patienten. Generelt kan nogle leukæmier være modtagelige for helbredelse med kemoterapi alene, men i dag skal der lægges stor vægt på såkaldte prognostiske faktorer, der er baseret på matematiske modeller, der gør det muligt for patienter at blive placeret i den grad af prognose, de har, og inkluderer:
1. Typen af leukæmi.
2. Den indledende molekylære ændring og dens persistens på trods af behandling eller udryddelse.
3. alder. Patienter over 60 år har en dårlig prognose sammenlignet med yngre patienter.
4. Kemoterapi. De angivne lægemidler og frem for alt de anbefalede doser bør anvendes. For eksempel i voksne LAL anvendelse af HyperCyVAD-ordningen (eskalerede doser cyclophosphamid, vincristin, adriamycin, deksamethason i kombination med cytosin arabinosid og methotreksat) opnår 90% CR og hærder i 50% af tilfældene, data, der ikke tidligere er set med andre ordninger. Denne behandling er giftig og kræver, at den kun bruges i institutioner, der har tilstrækkelige støtteressourcer. Desværre har ikke alle centre i vores miljø de anbefalede medicin, og resultaterne vil ikke være reproducerbare, da de ikke har tilstrækkelig støtte til patienten i fasen med maksimal myelosuppression.
5. Støttende terapi. Resultaterne af kemoterapi og nye lægemidler, nye kombinationer og med mere specificitet tvinger implementeringen af de tværfaglige hold med hæmatologens adresse; installation og anvendelse af centrale venøse katetre; støtte til blodbank til støtte for transfusioner af blodplader og erythrocytter (selv produkter stellat); infektologens intervention til påvisning af infektioner og passende anvendelse af antibiotika eller antifungale midler, enten profylaktisk eller terapeutisk, om nødvendigt; tilvejebringelse af isolationsrum med vedligeholdelses-og kvartmestertjenester, der gør det muligt at opnå et bakteriefrit miljø, herunder steril fodring; et laboratorium med patientprøvestyringsprotokol i specifikke behandlingsprotokoller og styring af specielle prøver (fremstilling af leukocytkoncentrater i buffy coat hematisk cytometri og opnå en optimal aflæsning); område forberedelse af højt specialiserede lægemidler, og på den anden side, det vigtigste er sygepleje, hjælpe-og administrativt personale, der med hele gruppen forventes de resultater, de har i andre lande.
6. Knoglemarvstransplantation (MO). Det er en kompleks og omkostningstype behandling, der kræver en kompatibel Mo-donor og en inaktiv tilstand med stor sandsynlighed for tidligt eller sent tilbagefald eller med faktorer med dårlig prognose. Det er en procedure med mere helbredende tendenser, da den bruger megadoser af kemoterapi til at udrydde leukæmiske celler, men i forsøget udrydder den også de normale forløbere, og udskiftning af en ny kompatibel normal marv er nødvendig. Typen allogen (søskendekompatibel identisk) er det bedste valg, da modtageren kun accepterer ved identitet delvis (den eneste af total accept er identiske tvillinger) og resulterer derfor i en afvisning af graft-versus-host-sygdommen, hvilket igen fører til graft-versus-leukæmi og øger udryddelsen af klonerne leukæmisk, men i alvorlige tilfælde af syndromet (grad 3-4) øges sygelighed og dødelighed på trods af den specifikke håndtering.
kroniske leukæmier
1. Kronisk lymfocytisk leukæmi. Det forekommer hyppigere hos ældre mennesker, og kriteriet er persistensen af lymfocytose på mere end 10 gange 109/l og MO med infiltration af mere end 50% lymfocytter med CD5+fænotype. Kriteriet for behandling er duplikering af kontoen for lymfocytter i et år eller progression af adenomegalia eller splenomegali, selv om nogle tilfælde ud over denne standard ved tilstedeværelsen af hæmolytisk anæmi eller trombocytopeni, autoimmun og derefter angivet behandlingen baseret på kombinationen af fludarabin, cyclophosphamid og prednison, i trin i og II kun somenter og observeret og forventet udvikling uden behandling.
2. Kronisk myeloid leukæmi (CML). I denne sygdom er der et stort fremskridt i kendskabet til tilstedeværelsen af Philadelphia-kromosomet, som blev beskrevet i 1950 i byen American Union, og at starten var det første markørkromosom i forbindelse med malignitet, men i løbet af årene med forskning lykkedes det os at kende t (9;22) med den funktionelle ekspression af kromosomet med produktion af et oncoprotein med stor thyrokinokinaseaktivitet, hvilket øger celleproliferationen, og som igen forklarer den store leukocytose og trombocytose, som disse patienter præsenterer, såvel som den store splenomegali (figur 5).
på dette tidspunkt er der gået 10 til 12 år siden opdagelsen af et lille molekyle specifikt rettet mod dette phosphat-donormolekylære substrat til den interne regulering af leukemicellen og dens substrater. Den oprindeligt kaldte STI (signaltransduktionsinhibitor) producerede konkurrencedygtig inhibering af phosphoryleringen, førte til celle apoptose, og patienterne opnåede kliniske resultater, der aldrig før blev set med molekylære remissioner på op til 80-90% efter 10 år, hvilket dramatisk ændrede sygdommens naturlige historie, med forbehandlingen var ikke mere end 3 år. Disse oplysninger gør en ændring i sygdommens naturlige historie og prognose, der tidligere var dødelig på kort sigt.

disse eksempler er en base for de fremskridt, der er så intense, at der opstod i de sidste par år, og at det er ønskeligt at tillade provokere interessen for medicinstuderende, deres lærere, men især for uddannelsesmyndighederne for leukæmi generelt, som optager de første 5 steder med hyppigheden af ondartet sygdom hos den voksne og de første steder hos børnene, så hæmatologien skal indgå i kernefagene i læseplanen for medicinens karriere og endda i de som en del af kurserne postgraduate specialisering.
selvom det er nyttigt for sin enkelhed, kan den britiske fransk-amerikanske klassifikation (FAB) føre til diagnostiske og derfor terapeutiske fejl i op til 20% af tilfældene. Af denne grund er klassificering efter immunhistokemi og molekylærbiologiske metoder blevet et absolut krav til korrekt klassificering og efterfølgende behandling af patienter (figur 6).

i modsætning til klassificeringen FAB afspejler hvem (tabel 2) en ændring i paradigmet, gennem hvilket vi forstår sygdomme blod, fordi for første gang kombineret den genetiske information, den morfologiske, cytokemiske og immunofenotypiske med de kliniske fund inden for de diagnostiske algoritmer af neoplasmer i det hæmatopoietiske væv; den relative betydning af hvert kriterium adskiller sig mellem neoplasmer, og der er ingen “guldstandard” til klassificering af alle hæmatologiske maligniteter. Målet var at definere enheder, der kunne anerkendes af patologer, og som havde klinisk relevans. Siden udseendet i 2001 er der foretaget forskellige revisioner for at opdatere indholdet i forhold til de nyeste opdagelser. Den sidste gennemgang var fra 2008 og klassificerer hæmatologiske maligniteter som følger:
1. Myeloide neoplasmer.
2. Lymfoide neoplasmer.
3. Mastcelle-eller fedtcellesygdomme.
4. Histiocytiske og dendritiske cellesygdomme.

NEOPLASIS MYELOID
myeloide neoplasmer er afledt af stamceller i knoglemarven, differentieres til erythrocytter, granulocytter (neutrofiler, basofiler og eosinofiler), monocytter og megakaryocytter. FAB-klassifikationen genkender 3 hovedkategorier:
1. Akut myeloid leukæmi.
2. Myelodysplastiske syndromer.
3. Myeloproliferative neoplasmer.
de vigtigste determinanter for kategorierne anerkendt ved hjælp af morfologiske, histokemiske og immunofenotypiske og procentdelen af blastceller, cellelinien og graden af differentiering af de neoplastiske celler (figur 7). I de senere år viste de genetiske egenskaber (cytogenetisk og molekylær) såvel som forbehandlingen og udviklingen af myelodysplasi en signifikant indflydelse på den kliniske opførsel af disse lidelser, som ikke altid korrelerer godt med kategorierne af FAB, så den centrale debat for omklassificering var at diskriminere mellem enhederne og patologiske prognostiske faktorer for at opnå en klassificering med klinisk relevans og betydning for patologen. Nogle genetiske abnormiteter synes at definere forskellige sygdomme, mens andre repræsenterer prognostiske faktorer for en bestemt sygdom.

i øjeblikket grupperer hvem klassificering myeloide tilstande i 4 hovedgrupper:
1. Myeloproliferative sygdomme
2. Myelodysplastiske syndromer
3. Myelodysplastiske / myeloproliferative sygdomme
4. Akutte myeloide leukæmier

Myeloproliferative sygdomme er en gruppe af klonale lidelser forbundet med proliferationen af en eller flere myeloide linjer. Det bliver mere og mere klart, at disse sygdomme ofte er forbundet med mutationer, der forårsager unormale stigninger i tyrosinkinaseaktivitet og vækstfaktoruafhængig progenitorcelleproliferation i knoglemarven. Procentdelen af blaster i knoglemarv er normal eller lidt høj, men altid mindre end 20%. Hæmatopoiesis er normalt effektiv, hvilket resulterer i en stigning i antallet af en eller flere modne celler i perifert blod. Prototypen af myeloproliferative neoplasmer er Philadelphia-kromosompositiv (Ph1) kronisk myeloid leukæmi (BCR/ABL). De øvrige inkluderede enheder er:
1. Polycythemia vera.
2. Idiopatisk myelofibrose.
3. Primær essentiel trombocytæmi.
4. Kronisk eosinofil leukæmi.
5. Kronisk neutrofil leukæmi.
6. Mastocytose.
7. Uklassificerbare myeloproliferative neoplasmer.

myelodysplastiske syndromer henviser til lidelser, der er kendetegnet ved ineffektiv celleproduktion og dysplasi, med en variabel risiko for transformation til akut leukæmi. Cellularitet i marven øges ofte, men meget variabel. Der er modning, men også dysplasi af en eller flere myeloide linjer. Hæmatopoiesis er ikke effektiv, og derfor eksisterer cytopenier. Denne vare indeholder:
1. Ildfast cytopeni med en-line dysplasi*.
• ildfast anæmi.
• refraktær neutropeni.
• refraktær trombocytopeni.
2. Ildfast anæmi med ringformede sideroblaster.
3. Ildfast cytopeni med dysplasi af flere slægter.
4. Ildfast anæmi med overskydende Blaster.
5. Myelodysplastisk syndrom med d (5K).
6. Uklassificerbart myelodyspastisk syndrom.
7. Juvenil myelodysplastisk syndrom inkluderer en foreløbig enhed kendt som juvenil refraktær cytopeni.
myelodysplastiske / myeloproliferative syndromer inkluderer lidelser, hvor dysplastiske og proliferative egenskaber eksisterer sammen. Denne gruppe omfatter myelomonocytisk juvenil leukæmi, som er repræsentativ for begge syndromer (myelodysplastisk og myeloproliferativ). Næsten halvdelen af patienterne har normale eller lave neutrofiltal og dysplasi af flere cellelinjer uden organomegali og knoglemarv med morfologi, der ligner ildfast anæmi med overskydende blaster, men med monocytose. Andre patienter har svær neutrofili, monocytose og splenomegali. Det vides endnu ikke, om de er 2 forskellige sygdomme, en myelodysplastisk og en myeloproliferativ; indtil videre er der ingen forskelle i cytogenetiske abnormiteter eller i vækstmønstrene i In vitro-kolonierne eller i deres kliniske udvikling, så der er kontrovers mellem klinikere og patologer i henhold til deres plads inden for klassificeringen. Ifølge den sidste revision er i denne kategori placeret:
1. Kronisk myelomonocytisk leukæmi.
2. Atypisk kronisk myeloid leukæmi (BCR/ABL negativ).
3. Juvenil myelomonocytisk leukæmi.
4. Myelodysplastisk / myeloproliferativt syndrom kan ikke klassificeres.
i kategorien af akutte leukæmier mielobl pursticas (LAM) (som er defineret af en procentdel større end 20% myeloblaster i knoglemarven eller blodfortyndende perifer eller tilstedeværelsen af en cytogenetisk abnormitet især på trods af blasternes konto) genkender følgende grupper:
1. LAM med tilbagevendende cytogenetiske translokationer.
2. LAM med myelodysplastiske egenskaber.
3. LAM og MDS relateret til antineoplastiske behandlinger.
4. LAM kan ikke klassificeres.
5. Myeloid sarkom.
6. Myeloide proliferationer relateret til ned syndrom.
7. Plasmacitoid blastisk neoplasma af dendritiske celler.
lymfoide neoplasmer
er dem, der stammer fra celler, der normalt udvikler sig til T-lymfocytter (cytotoksisk LT, samarbejdspartnere eller regulatorer) eller B-lymfocytter (lymfocytter eller plasmaceller). Generelt er lymfoide neoplasmer opdelt i dem, der stammer fra lymfoide forstadier og dem fra modne lymfocytter og plasmaceller og grupperes derefter efter deres afstamning (B eller T).
historisk set er lymfoide neoplasmer, der forekommer i knoglemarven og involverer knoglemarven, blevet adskilt fra dem, der præsenterer som en tumor (lymfom). Imidlertid er det nu kendt, at ethvert lymfom kan have kliniske træk ved leukæmi, og at enhver leukæmi lejlighedsvis kan forekomme som en tumor (granulocytisk sarkom). I hvem-klassificeringen afhænger diagnosen af flere lymfoide neoplasmer ikke kun af tumorcellernes anatomiske placering, men også på den morfologisk definerede Oprindelse af tumorcellen. Disse overvejelser ophævede relevansen af udtrykkene L1 og L2 i FAB-klassificeringen, da de ikke korrelerer med deres immunofenotype, genetiske abnormiteter eller med deres kliniske forløb (figur 8). L3 svarer til Burkitt lymfom i den leukæmiske fase og bør diagnosticeres som sådan.

1. Forstadier neoplasmer. Der er enighed om, at forstadier-neoplasmer, der præsenteres som faste tumorer, og dem, der involverer knoglemarv og blod, er biologisk den samme sygdom med forskellige kliniske præsentationer. De fleste forstadier lymfoide neoplasmer præsenteres som leukæmier, så det blev aftalt, at klassificeringen skulle bevare udtrykket LAL for den leukæmiske fase af type B og T forstadier neoplasmer. Der er 2 hovedkategorier:
• forløber B leukæmier / lymfomer.
• forløber leukæmier / lymfomer T.
2. Ældre B-celle neoplasmer. Den foreslåede klassificering betragter lymfomer og leukæmier af samme celletype som den samme sygdom med forskellige kliniske præsentationer eller stadier. Specifikke sygdomme afledt af modne B-celler er som følger:
1. Kronisk lymfocytisk leukæmi / lille lymfocytlymfom.
2. Lymfoplasmacytisk lymfom.
3. Mantelcellelymfom.
4. Prolymphocytisk B-celle leukæmi.
5. Follikulært lymfom.
6. Diffust storcellelymfom B.
• intravaskulært storcellelymfom B.
• primært mediastinalt storcellelymfom B.
• storcellelymfom B (relateret til Epstein Barr-virus-EBV).
•• histiocyt-og T-celle-rige store B-celle lymfom.
* diffust centralnervesystemet stort B-celle lymfom.
• * diffus primært kutant stort B-celle lymfom.
• diffus stor B-celle lymfom hos ældre positive for EBV.
• Pasmablastisk lymfom.
• primær pleural lymfom.
• * Alkoma-positiv (ALK) storcellet lymfom.
• Burkitt lymfom.
7. Marginal område B-celle lymfom.
8. Ekstranodalt marginalområde B-celle lymfom.
9. Miltens marginale område B-celle lymfom
10. Hår celle leukæmi.
11. Plasmocytom / plasmacelle myelom.

myeloide og lymfoide linje neoplasmer
nogle neoplasmer udtrykker markører af både myeloide og lymfoide linjer.udifferentieret) eller nuværende karakteristika for begge linjer (blandet fænotype eller blandet linje akut leukæmi).
tabel 3.Clasificaci de la OMS de las neoplasias mieloides y leucemias agudas
bibliografi til venstre
Bassan R, Hoels D. moderne terapi af akut lymfoblastisk leukæmi. J Clin Oncol. 2011;29:523-43. B. terapeutiske fremskridt i akut leukæmi. J Clin Oncol. 2011;29:487-94. Hr. formand, hr.formand, hr. formand for Rådet. 2008 hvem klassificering af lymfoide neoplasmer og videre: udviklende koncepter og praktiske anvendelser. Blod. 2011 12. maj;117 (19): 5019-32. Epub 2011 Februar 7. Anmeldelse. Cortes J, Hochhaus a, Hugues T, Kantajian H. Front Line og Bjærgningsterapier med tyrosinkinasehæmmere og andre behandlinger ved kronisk Myeloid leukæmi. J Clin Oncol. 2011;29:524-31.
Chin-Hon Pui, Carroll et al. Biologi, risikostratificering og terapi af pædiatrisk akut leukæmi: en opdatering. J Clin Oncol. 2011;29:551-65. multicenter uafhængig vurdering af voreskommer hos patienter med kronisk myeloid leukæmi behandlet med Imatinib. Tidsskrift National Cancer Institute. 2011;103:1-9.
Grever M, Lasse G. Moderne strategier for hår celle leukæmi. J Clin Oncol. 2011:29;583-90. Gribben JG, O ‘ Brien S. opdatering om behandling af kronisk lymfocytisk leukæmi. J Clin Oncol. 2011;29:544-50.
Hurtado MR, Vargas VP, Cortes FJ. Kronisk Myeloid Leukæmi. Aktuelle begreber i Fysiopatologi og behandling. Kræftkandidat. 2007:2:137-47.
Hurtado MR, Vargas VP, et al. Imatinib sammenlignet med Imatinib/cytarabin til førstelinjebehandling af tidlig Philadelphia-kromosompositiv kronisk Myeloid leukæmi. Resultater af et randomiseret klinisk forsøg med den Meksikanske samarbejdende Leukæmigruppe. Klinisk Leukæmi. 2008: 2(2);1128-32.
Lichtman MA. Klassificering og kliniske manifestationer af de klonale myeloide lidelser. Da: Thomas. Hæmatologi. Mc-Hill; 2010. Marcucci G, Haferlach T, Dohner H. Molekylær Genetik af voksen akut myeloid leukæmi. Prognostiske og terapeutiske konsekvenser. J Clin Oncol. 2011;29:475-86.
Rafael Hurtado M Mellado Y, Flores RG, Pablo Vargas. Semiolog for at få et godt tilbud. Rev Fac med UNAM. 2010; 53:36-43. m, Lo-Coco F. moderne tilgange til behandling af akut promyelocytisk leukæmi. J Clin Oncol. 2011;29:495-503.
Bemærk
* dysplasi. Det henviser til den cytomorfologiske ændring, der inkluderer dissociation af modning af kerne-cytoplasma (husk, at kromatinmodning afhænger af DNA og RNA-cytoplasma, derfor stopper kernen ved modning, mens cytoplasmaet fortsætter sin normale proces), der producerer ikke-levedygtige celler, og der er intramedullær apoptose.